Rivista Italiana di Genetica e Immunologia Pediatrica Italian Journal of Genetic and Pediatric Immunology Quadrimestrale di aggiornamento scientifico dell'Euromediterranean Paediatric Foundation
Esistono poi gli "Errori attivi" Per quanto riguarda gli errori attivi essi sono quelli pi๠facilmente individuabili in quanto fattori scatenanti dell'incidente. Si collocano a livello di persone e quindi il loro riscontro coincide spesso con l'identificazione di una responsabilità individuale. Le organizzazioni possiedono dei âsistemi di difesaâ che mirano proprio ad impedire il verificarsi degli incidenti.
Concentrando l'attenzione sugli incidenti in età pediatrica, secondo un'indagine effettuata dal National Health Service inglese , essi costituiscono il 6, 7 % di tutti gli incidenti segnalati. Secondo le stime emerse da tale studio, solo una piccola percentuale pari al 7%, si ਠverificata entro le mura domestiche o in strutture socio-sanitarie. Una percentuale molto pi๠ampia si ਠverificata in reparti di area critica, mentre il 10% si ਠverificata in ambito psichiatrico. Fortunatamente la maggior parte degli incidenti che coinvolgono neonati e bambini, non produce danni al paziente o, in molti casi, provoca danni di piccola entità . Gli errori statisticamente pi๠rappresentati variano a seconda della fascia d'età . Il 10% degli errori di terapia riguarda bambini tra 0 e 4 anni Pi๠frequenti per l'età pediatrica sono gli errori di terapia (17%), gli errori di trattamento o di procedura (13%) e gli errori di persona. Per i neonati sono pi๠frequenti gli errori di trattamento o di procedura (17%), gli errori di terapia (15%) e gli errori relativi all'accettazione, ai trasferimenti ed alla dimissione (14%). Alle origini degli eventi avversi ci sono molte possibili cause: la stanchezza dell'operatore, i difetti nella comunicazione tra le figure professionali (ad esempio una prescrizione illeggibile) o tra gli operatori ed il paziente, la mancanza di informazioni, la carenza di personale, il confezionamento dei farmaci (ad es. etichette quasi uguali per farmaci molto diversi), l'inidoneità di locali ed attrezzature, l'insufficiente addestramento. I bambini peraltro sono 3 volte pi๠esposti rispetto agli adulti ad errori terapeutici potenzialmente pericolosi.
Per ovviare a questi problemi si potrebbe intervenire tramite manovre preventive che prevedono ad esempio, tra le abitudini mediche, ad inserire nelle prescrizioni peso ed eventuali allergie del paziente, ad abolire abbreviazioni ed indicazioni generiche o quantomeno standardizzarle. Altra mossa preventiva sarebbe quella di attivare la prescrizione medica computerizzata con software che supporta le decisioni cliniche (rischi allergici, dosi, frequenza di somministrazione).
Andando ad indagare quali siano gli elementi che permettono ai bambini ed alle loro famiglie di sentirsi oggetto di cure sicure, sono emersi i seguenti dati: il linguaggio usato dal personale socio-sanitario ਠun elemento fondamentale nel rassicurare i giovani pazienti. Parlare al bambino con un linguaggio adeguato all'età e fornirgli spiegazioni complete ed esaurienti circa la diagnosi, il percorso terapeutico ed i trattamenti farmacologici a cui verrà sottoposto gli permette di sentirsi sicuro dell'efficacia delle cure. Le famiglie straniere necessiterebbero dell'effetto rassicurante di ricevere le informazioni circa la salute del bambino nella propria lingua. I genitori spesso non si sentono ascoltati dai professionisti sanitari quando hanno esposto le condizioni cliniche del figlio o l'aggravarsi della patologia di base; in queste circostanze i genitori potrebbero ritenere che i provvedimenti presi dai medici e dagli altri professionisti siano insufficienti o inadeguati. Di fondamentale importanza per evitare rischi clinici, si indicano ancora la sicurezza dell'ambiente ed il percepire che i professionisti sanitari sono vigili nel prevenire situazioni pericolose. Merita menzione il consenso informato, cardine della gestione del rischio clinico, non tanto nella consueta chiave di lettura giuridica (validità del consenso in relazione all'età , alle condizioni psicofisiche ecc.), quanto come fondamentale processo di comunicazione, nel quale il medico si gioca ampia parte della fiducia del paziente. Ciಠha notevole importanza nel prevenire azioni rivendicative, soprattutto allorquando si verifica un evento avverso. Per superare le criticità relative al momento della dimissione si dovrebbe evitare di fornire informazioni parziali, frettolose, scarsamente comprensibili, circa l'assistenza domiciliare e l'eventuale prosieguo delle cure, tenendo anche conto di quelle minoranze etniche da cui ci separano barriere linguistiche e culturali. à necessario quindi promuovere un atteggiamento attivo molto ampio, di vasti orizzonti, nella ricerca delle possibili fonti di eventi avversi per migliorare ulteriormente la sicurezza dei pazienti in età pediatrica. Il messaggio saliente che vorremmo emergesse ਠche tutte le figure professionali devono impegnarsi per evitare che si verifichino delle condizioni potenzialmente lesive per il paziente. Il medico, l'infermiere o il manager della sanità , ma anche il biologo o il farmacista sono chiamati, a livelli diversi, a prendere decisioni che possono generare criticità ed innescare conflitti. Essi sono impegnati in una continua gestione di contrasti, in cui appare preponderante il saper generare consenso piuttosto che avere potere decisionale. Per creare consenso ਠnecessario riconoscere, comprendere e neutralizzare gli eventi e le percezioni che possono generare il conflitto, o aggravarlo, rendendolo difficilmente gestibile. Il conflitto puಠessere utilmente evitato o governato attraverso l'utilizzo di tecniche di negoziazione relazionale, idonee a ridurre la criticità . Proprio la riduzione della criticità , e degli eventi a essa connessi, determina la diminuzione del danno iatrogeno, delle sofferenze inutili e delle morti evitabili che significativamente occorrono nella cura della salute. La negoziazione relazionale quindi ਠl'unica metodica capace di ridurre il rischio clinico creando valore: gestionale, economico, umano. Dalla possibilità di sviluppo di errore non ਠscevra la ricerca intesa come un'attività che si basa su regole precise che consentono di arrivare, attraverso percorsi ben definiti, a un risultato concreto, oggettivo e riproducibile: in poche parole si basa sul metodo scientifico, lo stesso introdotto nel XVI-XVII secolo da Galileo Galilei, considerato il padre della scienza moderna. Questi concetti risultano calzanti anche per la ricerca applicata o translazionale o, come si usa dire recentemente di ricerca " from bench to bedside", âdal bancone di laboratorio al letto del paziente". In realtà questo rapporto ਠbidirezionale: se da un lato il paziente, soprattutto quello con malattia rara su base genetica, ci insegna a comprendere meglio i meccanismi di funzionamento del nostro organismo (from bedside to bench), dall'altro le conoscenze che acquisiamo possono essere utilizzate per migliorare la diagnostica e la cura dei pazienti (from bench to bedside). Si tratta, in altre parole, di costruire una sorta di ponte tra la scienza e la medicina, per poter utilizzare nel modo migliore le scoperte dei ricercatori, un ponte che sia a due sensi di marcia. Il percorso tradizionale prevede che le informazioni che arrivano dal laboratorio vengano tradotte in strumenti utili da applicare al letto del paziente, cioਠalla pratica clinica di tutti i giorni, ma non ਠraro che da informazioni che arrivano dall'osservazione dei pazienti i ricercatori colgano spunti per nuovi esperimenti in laboratorio. Il concetto non ਠnuovo, ma ha assunto un significato molto diverso negli ultimi anni: fin dalla metà del secolo scorso esisteva uno stretto legame tra ricerca di base e medicina, ma oggi le due discipline viaggiano a velocità molto diverse. La ricerca di base ਠincredibilmente veloce, produce risultati a ritmi molto rapidi, mentre i tempi per portare questi risultati al letto del paziente sono molto lunghi e spesso accade che le enormi possibilità nella diagnosi o nella terapia suggerite dalla scienza non possano essere sfruttate fino in fondo dai medici che si confrontano ogni giorno con i pazienti.
Serve dunque che il medico diventi un esperto capace di tradurre in pratica le scoperte della scienza, cercando le strategie migliori per poter utilizzare âsul letto del pazienteâ l'ultima scoperta nel campo della genetica o della biologia molecolare. Ed ਠper questo che nei laboratori pi๠avanzati nascono centri specifici di ricerca traslazionale presso i quali lavorano persone capaci di camminare in equilibrio tra i due ambiti: si tratta di ricercatori che conoscono la ricerca di base, ma che hanno anche un'attenzione particolare per il paziente e una grande capacità di comprendere le necessità di chi si prende cura dei malati ogni giorno nella pratica clinica. Ci auguriamo quindi che , con animo umile, sensibile ed attento, possiamo crescere in questa nuova realtà , con questi nuovi orizzonti, tutelando i nostri pazienti, tutelando al tempo stesso la nostra professionalità .
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Telemedicina con le punte delle dita
Giuseppe Micali
Funzionano con una rete Wifi o con una mini-SIM card, consentono l'accesso ad internet scegliendo un piano tariffario dai pi๠famosi gestori di telefonia mobile e sono divenuti uno strumento di utilità collettiva. Sono l'iPhone e l'iPad e si stanno utilizzando sempre pi๠come strumenti per il medico per comunicare in tempo reale e fornire l'assistenza clinica necessaria in emergenza. à, percià², possibile effettuare una visita medica a distanza grazie al software FaceTime, che consente la videochiamata in modo semplice ed istantaneo. Il primo progetto sperimentale di telemedicina con il tablet iPad si sta svolgendo all'Ospedale Niguarda di Milano e al Policlinico Gemelli di Roma dove i medici possono accedere, ovunque si trovino, alla consultazione della cartella clinica elettronica, agli esami diagnostici e alle immagini radiologiche del paziente. Inoltre, un sistema di prevenzione sanitaria, "Healthpresence" consentirà al medico di visitare un paziente a distanza. Siamo arrivati alla telemedicina con le punte delle dita, in grado di ascoltare il battito di un cuore o di misurare la pressione in tempo reale e ci siamo anche accorti che la fantascienza ਠappena incominciata. Adesso ਠmeglio far parlare i links.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
La cute come organo immune Skin as immune organ
G. L. Marseglia, A. Licari Dipartimento di Scienze Pediatriche e Patologia Umana ed Ereditaria, Università degli Studi di Pavia, Fondazione IRCCS Policlinico San Matteo Pavia
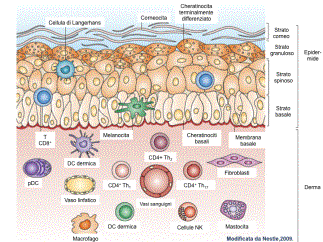
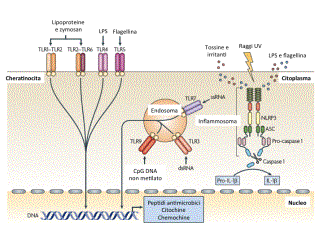
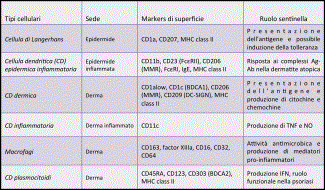
La cute presenta numerose attività funzionali. Una delle pi๠importanti ਠla funzione di barriera di protezione nei confronti dell'ambiente esterno, garantita dai cheratinociti epidermici; il loro continuo turn-over cellulare assicura il mantenimento dell'integrità dell'epidermide e costituisce un efficiente ostacolo in grado di limitare anche la perdita di acqua e di elettroliti verso l'esterno. La presenza dei melanociti epidermici assicura la funzione di difesa nei confronti delle radiazioni ultraviolette (UV) ; la matrice extracellulare del derma conferisce l'attività di tensione elastica; inoltre la cute costituisce l'organo effettore principale del mantenimento dell'omeostasi termica e dell'equilibrio idro-elettrolitico; infine, attraverso la rete delle terminazioni nervose, consente le fondamentali attività sensoriale, conoscitiva e di comunicazione. Recentemente ਠstata definita una nuova attività funzionale, cioਠquella immunologica. La sorveglianza di un organo di cosଠgrandi dimensioni e cosଠesposto all'ambiente rappresenta una sfida unica per le cellule sentinelle ed effettrici del sistema immune. La cute, infatti, puಠdare inizio a risposte infiammatorie e immunitarie verso sostanze potenzialmente nocive che vengono a contatto con essa per penetrazione dall'esterno (proteine batteriche, micetiche, virali, apteni) o per diffusione ematogena da altri distretti dell'organismo o formatisi al suo interno (prodotti del metabolismo di alcuni farmaci). I principali protagonisti di questo scenario sono i cheratinociti, le cellule di Langerhans, le cellule dendritiche cutanee, i linfociti cutanei, i mastociti e le cellule endoteliali (Fig.1) ; queste popolazioni cellulari, tra loro integrate e correlate nel cosiddetto âsistema immunitario cutaneoâ, possono esercitare effetti biologici non solo a livello cutaneo ma anche a distanza. I cheratinociti, che costituiscono la popolazione cellulare epidermica numericamente pi๠rappresentata, sono in grado non solo di sintetizzare i principali costituenti molecolari della barriera epidermica, ma anche di intervenire attivamente nella regolazione delle reazioni infiammatorie immunomediate della cute: possono infatti esprimere molecole MHC di I classe e, in particolari condizioni di attivazione, anche molecole di II classe e alcune molecole di adesione come LFA-3 (Leukocyte Functional Antigen 3) e ICAM-1 (Intercellular Adhesione molecule 1) che interagendo con i loro ligandi naturali sui leucociti e producendo una moltitudine di chemochine, fattori di crescita e citochine richiamano nell'epidermide cellule dendritiche, linfociti T e altri leucociti; possono liberare fattori in grado di modulare la funzione delle cellule di Langerhans, oltre che l'accumulo, la sopravvivenza e l'attivazione di linfociti T nella cute; esprimono inoltre sulla loro membrana i TLR (Toll-Like Receptors), soprattutto di tipo 1, 2 e 5 che hanno un ruolo determinante nell'immunità innata in quanto, essendo in grado di riconoscere sequenze specifiche espresse dalla maggior parte dei microrganismi patogeni, attivano la produzione di peptidi antimicrobici (tra cui beta-defensine 1-3 e catelecidine LL-37) e di citochine e chemochine, come IL-8, considerata il pi๠potente fattore chemotattico per i granulociti neutrofili. Da queste considerazioni emerge quindi l'importanza del ruolo dei cheratinociti come âsensori del pericoloâ in grado di discriminare tra organismi innocui e agenti patogeni dannosi e successivamente di attivare la risposta immunitaria attraverso diversi sistemi di allarme, ad esempio attraverso l'espressione dei TLR o attraverso il complesso proteico dell'inflammasoma (Fig.2). Le cellule dendritiche svolgono il ruolo chiave di âsentinellaâ essendo strategicamente posizionate in distinti compartimenti anatomici cutanei; ciascun tipo cellulare possiede inoltre specifiche proprietà funzionali, come ad esempio la secrezione di mediatori pro-infiammatori, la produzione di IFN-1 o la presentazione dell'antigene. Le cellule dendritiche della cute e le cellule di Langerhans hanno una funzione essenziale nell'induzione delle risposte immunitarie primarie, essendo in grado di presentare l'antigene ai linfociti T e di attivare quindi la risposta linfocitaria nei confronti sia di neo-antigeni cutanei che di antigeni già riconosciuti, inibire una risposta immune parzialmente dannosa nei confronti di antigeni cutanei non pericolosi e attivare l'immunità naturale attraverso l'interazione tra peptidi microbici e TLR (Fig.3). Nella cute umana normale sono presenti numerosi linfociti che appartengono prevalentemente al fenotipo T (linfociti T cute-specifici) e sono localizzati soprattutto in sede perivascolare nel derma (98%) e solo in piccola quantità nell'epidermide (2%) ; la caratterizzazione immunofenotipica di queste cellule ha permesso di rilevare che la maggior parte di esse sono da considerarsi cellule memoria. Di recente identificazione ਠla presenza della linea cellulare T effettrice Th17 presente anche a livello cutaneo; questa popolazione cellulare, distinta dai sottotipi Th1 e Th2, ਠresponsabile della produzione di citochine infiammatorie (IL-17A, IL-17F, IL-22, e IL-26) coinvolte in diversi modelli di malattie autoimmuni, come la sclerosi multipla, l'artrite reumatoide, le malattie infiammatorie croniche intestinali e la psoriasi, cosଠcome nella risposta immune ai patogeni extracellulari (es. C. albicans, M. tuberculosis). Le cellule Th17 hanno inoltre un ruolo nella patogenesi di alcune malattie allergiche; recentemente ਠstata dimostrata la presenza dei Th17 e dell'IL-17 nei pazienti con dermatite atopica: la polarizzazione Th17 indotta dagli eosinofili sosterrebbe non solo la fase acuta della malattia ma sarebbe necessaria per il passaggio alla fase cronica, attraverso l'attivazione dei processi di rimodellamento cutaneo stimolati da citochine proinfiammatorie come IL-8, IL-6, e IL-11. Recenti evidenze hanno messo in luce una nuova popolazione di linfociti T, definiti ânon convenzionaliâ e rappresentati dalle cellule Tγδ e dalle cellule iNKT, che sembrano entrambe coinvolte nei processi di immunosorveglianza cutanea, essendo in grado di esercitare attività citolitica ed apoptosica diretta nei confronti di cellule infette o trasformate. Inoltre le cellule Tγδ ï producono fattori di crescita indispensabili per la guarigione delle ferite (Fig.4). Le cellule iNKT sono state inoltre nella patogenesi dell'asma; controverso ਠinvece il loro ruolo nella patogenesi della dermatite atopica in cui sembrerebbero giocare un ruolo essenziale nella risposta immune allergica innata: in particolare, la loro presenza sembrerebbe correlare con la gravità delle lesioni cutanee caratteristiche della malattia. I mastociti, dislocati prevalentemente in sede perivascolare, svolgono un ruolo altrettanto rilevante nella regolazione delle reazioni immunoflogistiche acute e croniche; essi, infatti, contengono nei loro granuli citochine preformate in grado di attivare le cellule endoteliali e di modulare la differenziazione dei linfociti T. Inoltre, una volta attivati, i mastociti sintetizzano chemochine e citochine che promuovono il reclutamento e l'attivazione di vari leucociti; infine, essi contraggono stretti rapporti anatomici e funzionali con le terminazioni nervose peptidergiche che, oltre a veicolare informazioni nocicettive, sono in grado di rilasciare perifericamente peptidi neuroregolatori (sostanza P, VIP, CGRP ovvero calcitonin gene-related peptide) con importanti funzioni nell'induzione e regolazione della flogosi. Le cellule endoteliali, che rivestono la superficie interna dei vasi dermici, sono provviste di numerose attività biologiche, le pi๠importanti delle quali sono la sintesi e la secrezione all'esterno di numerose molecole: componenti della matrice extracellulare, fattori della coagulazione, sostanze ad attività vasodilatante, fattori di crescita e mitogeni cellulari, citochine, chemochine e molecole di adesione; in condizioni di attivazione, inoltre, esse sono in grado di esprimere sulla loro membrana molecole di classe II e recettori per chemochine, per l'Fc delle IgG e per il C3 del complemento. Le cellule endoteliali, infine, costituiscono una barriera selettiva in grado di regolare il traffico cellulare tra torrente circolatorio e tessuti attraverso l'espressione sulla loro superficie di numerosi recettori, come quelli che possono riconoscere gli homing receptors dei linfociti, oppure le molecole di adesione presenti sulla membrana dei granulociti e dei monociti. In conclusione, le varie popolazioni cellulari che compongono il sistema immunitario cutaneo costituiscono una vera e propria unità integrata e interattiva, che da un lato mette in relazione l'organismo con l'ambiente esterno e dall'altro deve fornire una protezione adeguata nei confronti di agenti ambientali potenzialmente dannosi. Un'alterazione persistente, per motivi genetici e/o acquisiti, della regolazione reciproca di tali componenti ਠalla base di diverse patologie della cute che trovano estrinsecazione anche in età pediatrica.
Figura 1 - Anatomia della cute ed effettori cellulari, modificata da Nestle, 2009 La struttura della cute riflette la complessità delle sue molteplici funzioni: essa svolge infatti il ruolo di barriera protettiva, ਠcoinvolta nel mantenimento della temperatura corporea, nel raccogliere e trasmettere informazioni relative all'ambiente esterno e ha un ruolo attivo nel sistema immune. L'epidermide contiene lo strato basale, lo strato spinoso, lo stato granuloso e lo strato corneo, che ਠresponsabile della funzione di barriera della cute. Le cellule specializzate dell'epidermide sono rappresentate dai melanociti e dalle cellule di Langerhans. Qualche cellula T, principalmente linfociti T CD8+ citotossici, ਠpresente nello strato basale e nello strato spinoso. Il derma ਠcomposto da collagene, fibre elastiche e fibre reticolari e contiene molte cellule specializzate, come le cellule dendritiche (DC) nei vari sottotipi, incluse le DC dermiche e le DC plasmocitoidi (pDC), e i linfociti T, compresi i T helper CD4+ (Th1), Th2 e Th17, le cellule Tγδ e le iNK T (invariant NK T). Sono inoltre presenti macrofagi, mastociti e fibroblasti. I vasi sanguigni e linfatici e le fibre nervose sono inoltre presenti in tutto il derma.
Figura 2 - Cheratinociti come sensori del pericolo, modificata da Nestle, 2009 I cheratinociti svolgono il ruolo di âsensori del pericoloâ in quanto in grado di riconoscere agenti esterni e potenzialmente dannosi (come ad esempio il Lipopolisaccaride (LPS) batterico, le tossine, gli irritanti e i raggi UV) attraverso i Toll-Like Receptors (TLRs) e il complesso dell'inflammosoma. I TLRs sono recettori transmembranari presenti sulla superficie cellulare o sulla superficie del compartimento endosomiale. Il LPS stimola il TLR4; le lipoproteine batteriche e lo zymosan fungino i complessi eterodimerici formati da TLR1-TLR2 e TLR2-TLR6; la flagellina batterica attiva il TLR5; le triplette CpG non metilate presenti nel DNA funzionano come stimolatori del TLR9 endosomiale; la doppia elica RNA (dsRNA) attiva il TLR3 endosomiale; la singola elica RNA (ssRNA) attiva il TLR7. Il riconoscimento di agenti esterni attiva un meccanismo che attraverso l'attivazione di segnali cellulari culmina nella risposta immune innata ed adattativa con produzione di peptidi antimicrobici, citochine e chemochine. I cheratinociti inoltre esprimono la proteina NLRP3, che appartiene alla classe NLR (nucleotide-binding domain, leucine-rich repeat-containing), di recente identificazione , che sarebbe in grado di attivare una risposta nei confronti degli agenti esterni a livello citoplasmatico e di attivare il complesso dell'inflammosoma. Quest'ultimo rappresenta un complesso multimerico formato da una proteina NLR, da una proteina adattatrice chiamata ASC (apoptosis-associated speck-like protein) e dalla pro-caspasi 1; la sua attivazione termina nella produzione di caspasi 1 , che trasforma a sua volta a pro-IL-1ï¢ in IL-1ï¢ï biologicamente attiva.
Figura 3 - Cellule dendritiche e macrofagi, modificata da Nestle, 2009 Le cellule dendritiche (CD) cutanee possono essere classificate in base alla loro localizzazione in distinti compartimenti anatomici: le cellule di Langerhans sono il sottotipo cellulare maggiormente rappresentato nell'epidermide, dove sono costitutivamente residenti insieme ai cheratinociti; le cellule dendritiche dermiche risiedono immediatamente al di sotto della giunzione dermo-epidermica e in tutto il compartimento dermico. In base alla loro localizzazione anatomica, i diversi tipi cellulari hanno specifiche prorpietà funzionali, come la secrezione di citochine pro-infiammatorie (da parte delle CD infiammatorie), di IFN-1 (da parte delle CD plasmocitoidi) o la presentazione dell'antigene a livello cutaneo.
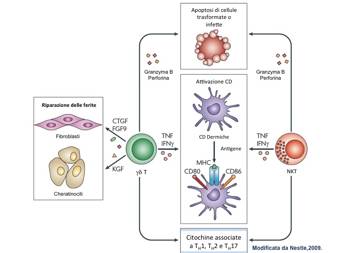
Figura 4 - Cellule T non convenzionali, modificata da Nestle, 2009 I linfociti T non convenzionali, come le cellule γδ T e le iNKT (invariant natural killer T) sono coinvolte nell'immunosorveglianza cutanea. Entrambi i tipi cellulari hanno proprietà citolitiche e rilasciano granzyma B e perforina e causano apoptosi di cellule infette o trasformate. Esse sono in grado di attivare le cellule dendritiche attraverso la produzione di TNF e IFN-γ. In pi๠le cellule γδ T producono fattori di crescita che sono essenziali per la riparazione delle ferite , come il fattore di crescita del tessuto connettivo (CTGF), il fattore di crescita dei 9 (FGF9) e il fattore di crescita dei cheratinociti (KGF). Infine, entrambi i tipi cellulari, Tγδ e iNKT, producono citochine che sono di solito associati ai fenotipi T helper 1 (TH1), TH2 e TH17.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Immunogenetic classification of immune mediated disease
Tradotto e curato da: Sara Manti, Valeria Ferraà¹, Donatella Comito, Claudio Romano Dipartimento Scienze Pediatriche, UOC Genetica ed Immunologia Pediatrica, Università di Messina
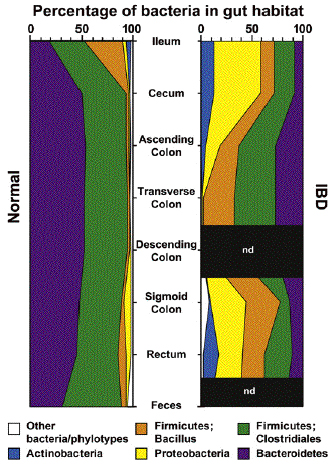
Non trascurabile ਠla componente immunologica-ambientale. Ruolo chiave sembra essere svolto dalla flora batterica, che risente fortemente di disregolazioni geniche, ma anche di fattori ambientali come il tipo di alimentazione, additivi, stress, fumo, sostanze chimiche (farmaci: FANS). Sembra che una condizione di disbiosi intesa come alterazione della composizione del microbiota possa essere considerato un primum movens nella genesi di una alterata risposta immune a livello intestinale. Nel soggetto normale, infatti, il microbiota ਠcostituito da Bacteroidetes, Firmicutes e Clostridiales; questi componenti sono presenti in quantità ridotta sulla superfice mucosale in un soggetto con IBD, ove invece prevalgono Firmicutes Bacillus, Proteobacteria ed Actinobacteria.
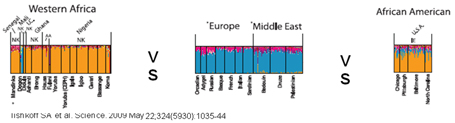
Numerosi sono i geni candidati al determinismo delle IBD. I cromosomi su cui mappano sono 1 (TNF-R, HSPG2, UBE1L, TGF-β2, TGF-β4, E2G), 3 (CCR5, GNAI2, hMLH1, IL12A) , 4 (IL-2), 5 (CSF-2, IL3-5.2), 6 (HLA I-III, MICA, NFκB, IFN-γ R), 7 (MUC 3, EGFR, HGF) , 10 (β1-integrin), 12 (MUC 3, EGFR, HGF), 14 (TCR α and δ, Proteasome cluster, Leucotriene B4R) , 16 (IL-4R, CD10, CD11, E-caderina) , 19 (ICAM1, C3, TBXA2, LTB4H). Le analisi di associazione genetica, tramite lo studio del genoma completo, hanno identificato numerosi loci di suscettibilità , designati con le sigle da IBD1 a IBD6, rispettivamente sui cromosomi 16, 12, 6, 14, 5 e 19. Il locus IBD1, che ha mostrato la maggiore associazione, viene denominato NOD2/CARD15. Mentre il possesso di una copia dell'allele determina un piccolo aumento del rischio di sviluppare il MC (da 2 a 4 volte), il possesso di 2 copie (in omozigosi o eterozigosi composta) lo aumenta di 20-40 volte. Queste varianti genetiche alterano l'attivazione del fattore nucleare NF-kb intestinale in risposta ai lipopolisaccaridi e ai peptidoglicani batterici. Altri loci IBD possono codificare per il gene della resistenza multi-farmaco e per i cluster dei geni trasportatori di cationi organici. Solo il 20% di pazienti con MC ਠomozigote per le varianti di NOD2. Il possesso degli alleli a rischio NOD2/CARD15 ਠassociato con i fenotipi ileale e stenosante. Questi geni sono rari nei pazienti afroamericani, mentre sono presenti nel 40-50% dei pazienti caucasici. Grazie al GWAS, un ampio studio di associazione sul genoma, noto anche come complesso studio di associazione sull'intero genoma (studio WGA, o WGAS) ਠstato effettuato l'esame di tutti o la maggior parte dei geni di diversi individui per valutarne il grado di espressività . Il genoma umano contiene molti milioni di polimorfismi a singolo nucleotide, e altre migliaia di variazioni nel numero di copie di piccoli e grandi segmenti del genoma, che possono causare cambiamenti del fenotipo ed influenzare le variazioni individuali e la suscettibilità alle malattie. Studi GWAS consentono ai ricercatori di analizzare campioni di 500.000 o pi๠SNPs per ogni soggetto. Ad oggi, questi studi, hanno identificato ben >30 fattori di rischio e mappato i geni coinvolti nell'insorgenza delle IBD in età pediatrica ed adulta. Le mutazioni geniche recentemente individuate sono le seguenti: - 5p13.1 crom.: modula l'azione del PTGER4, proteina appartenente alla famiglia dei recettori accoppiati alla G-proteina. Trattasi di uno dei quattro recettori individuati per prostaglandina E2 (PGE2). Essa funge da segnale di attivazione dei linfociti T; influenza l'espressione di PGE2 e la rapida crescita cellulare in risposta all'EGR1; regola il livello e la stabilità della cicloossigenasi-2 mRNA; porta alla fosforilazione della glicogeno-sintasi/chinasi-3. - à stato ipotizzato che la stratificazione delle IBD in base all'età di esordio potrebbe identificare geni aggiuntivi associati con la flogosi intestinale. A tal fine, ਠstata effettuata un'analisi GWAS in una coorte di 1.011 individui con malattia infiammatoria intestinale ad esordio pediatrico e confrontati con 4.250 controlli. Sono stati cosଠidentificati e replicati geni significativamente associati, non noti in precedenza, sui loci dei cromosomi 20q13 e 21q22, situati, rispettivamente, vicino al TNFRSF6B e PSMG1. - SMAD3: fattore trascrizionale che regola l'espressione del TGF-β, a sua volta coinvolto nell'induzione di FOXp3. La carenza di SMAD3e/o la sua ridotta fosforilazione inducono una differenziazione dei T-naive in senso Th17 con una maggiore predisposizione allo sviluppo di malattie autoimmuni. - TNFSF11 (RANKL) : coinvolto nell'attivazione dell' NF-kb, stimola le cellule dendritiche, la proliferazione dei linfociti T-naive e loro differenziazione in Treg. I valori di tale fattore trascrizionale sono aumentati nel plasma di pazienti affetti da MC. - IL10RA IL10RB: lo studio ਠpartito da un'analisi dei collegamenti genetici e del sequenziamento genomico di due famiglie di soggetti con bambini affetti da malattie infiammatorie intestinali. Da questi studi ਠemerso che in quattro pazienti su nove con una colite precoce sono stati identificati tre distinti mutazioni omozigotiche nei geni IL10RA e IL10RB, che codificano rispettivamente per IL10R1e IL10R2. Queste stesse proteine sono responsabili della formazione del recettore interluchina-10. Di conseguenza, i segnali di quest'ultima vengono inibiti inducendo un'aumentata secrezione di fattori di necrosi tumorale e altre citochine pro-infiammatorie: l'intero sistema immunitario risulta alterato. - NCF4: la proteina codificata da questo gene ਠun componente citosolico necessario per la produzione, da parte dei fagociti, di NADPH-ossidasi, enzima importante per la difesa ospite. Questa proteina ਠpreferenzialmente espressa nelle cellule della linea mieloide. Interagisce principalmente con NCF2/p67-phox per formare un complesso con NCF1/p47-phox ed G-RAC1. Questo complesso attiva quindi il β-flavocitocromo, centro integrato di membrana catalitica del sistema enzimatico. Il dominio PX di questa proteina puಠlegare i prodotti fosfolipidi della 3-PI-chinasi. La fosforilazione di questa proteina regola negativamente l'attività dell'enzima. Le mutazioni individuate a carico di tale gene predispongono allo sviluppo di febbre, diarrea, dolori addominale, eczema, sinusite, croup e colite granulomatosa. Studi associati-Genoma (GWAS) hanno identificato un numero rapidamente crescente di varianti genetiche, circa 80, potenzialmente associabili al rischio di IBD. Questi dati recenti ultime potrebbero fornire, inoltre, la possibilità di studiare meccanismi predisponenti la malattia ed individualizzare un adeguato approccio terapeutico. I risultati hanno evidenziato l'importanza della relazione tra la risposta ai microbi intracellulari (autofagia, fagocitosi, funzione di barriera) ed immunità innata e adattativa (cellule Th17, T-reg) nella patogenesi delle IBD. Con i rapidi progressi nella genetica umana, ਠdiventato chiaro che una delle principali sfide per lo studio dei caratteri genetici ਠquello di determinare come i geni della malattia e dei loro alleli corrispondenti esercitino la loro influenza sulla biologia di salute e malattia, e come applicare la biologia al fine di prevenire la malattia stessa. à altresଠvero che terapie pi๠efficaci (comprese immunomodulatori e biologici), sono stati sempre pi๠utilizzate per il trattamento di pazienti con IBD. Recentemente, il concetto di "top down" nell'approccio terapeutico suggerisce che, se queste classi di farmaci sono utilizzati all'esordio di malattia possono modificare la storia naturale di malattia con riduzione del rischio di complicanze. Tuttavia, le terapie pi๠recenti sono costose, dotate di un aumentato rischio di infezione e di linfoma, e non efficaci su tutta la popolazione affetta. Pertanto, lo sviluppo e validazione di biomarkers di malattia in grado di distinguere fenotipi ad âaltoâ ed a âbassoâ rischio, appaiono necessari. Non si tratta di una novità ma di un approccio clinico-laboratoristico già utilizzato in altre branche mediche quali l'oncologia. Sarebbe, inoltre, opportuno analizzare popolazioni non caucasiche, giacchਠle varianti geniche afro-americane sono ben diverse da quelle caucasiche. Pertanto possono esser utili in tal senso studio condotti su gruppi familiari e su coorte.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Review
Ossigenoterapia
Rossella Pecoraro, Tiziana Timpanaro, Papale Maria, Francesco Di Mauro1 Dipartimento di Pediatria, Università degli studi di Catania, 1Dipartimento di Pediatria 2 Università Di Napoli
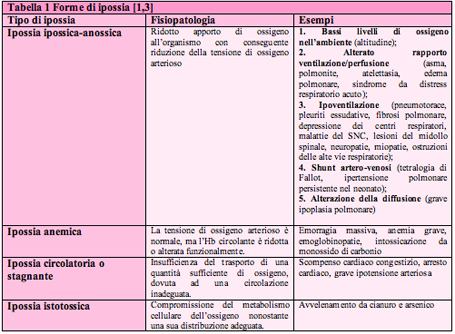
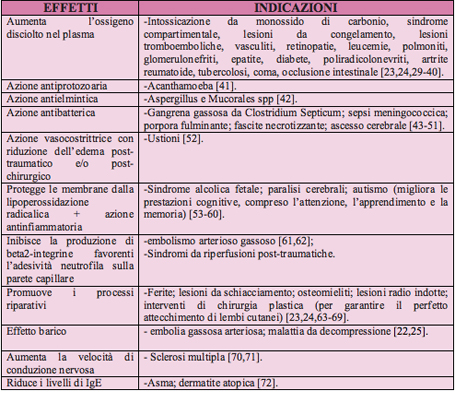
Definizione L'ossigenoterapia consiste nella somministrazione di ossigeno, generalmente miscelato con aria, in circostanze patologiche che impediscono la normale ossigenazione del sangue e dei tessuti. Lo scopo dell'ossigenoterapia ਠquello di evitare l'istaurarsi dell'ipossia, aumentando la concentrazione dell'ossigeno, e quindi la sua tensione parziale negli alveoli polmonari, in modo da favorirne il passaggio dallo spazio alveolare al sangue [1]. Basi di fisiopatologia respiratoria L'insufficienza respiratoria rappresenta la principale causa di ossigenoterapia e si definisce come l'incapacità dei polmoni a soddisfare le esigenze metaboliche dell'organismo. Si verifica per riduzione della capacità del sistema respiratorio a mantenere l'omeostasi degli scambi gassosi ed ਠcaratterizzata dalla presenza di una PaO2 <60 mmHg o di una PaCO2 >50 mmHg [2]. L'alterazione pi๠comunemente riscontrata in corso di insufficienza respiratoria ਠla diminuizione della concentrazione di ossigeno nel sangue arterioso (ipossiemia), cui puಠfar seguito una anomala ossigenazione tissutale (ipossia), associata talvolta ad una ridotta eliminazione di anidride carbonica (ipercapnia). Si distinguono quattro forme di ipossia che sono riassunte nella tabella 1 [1].
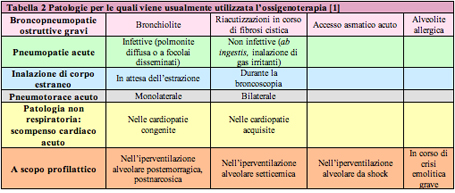
Al fine di definire una corretta indicazione all'ossigenoterapia ਠnecessario distinguere l'insufficienza respiratoria in due differenti forme [tabella 2]: ⢠Insufficienza respiratoria di tipo I: definita ipossiemico e normo/ipocapnica. E' relativa a patologie delle vie aeree centrali (croup, corpo estraneo, anafilassi, tracheite/epiglottite batterica, ascesso retrofaringeo) e del parenchima polmonare (asma, bronchiolite, polmonite, edema polmonare, fibrosi cistica, displasia broncopolmonare). E' determinata da alterazioni del rapporto ventilazione/perfusione (V/Q) con persistenza di una buona perfusione in aree del polmone poco ventilate (accesso acuto d'asma, bronchiolite, malattia delle membrane ialine nel neonato) od anche da condizioni che riducono la perfusione polmonare con ventilazione conservata (embolia polmonare, cardiopatia congenita cianotica, scompenso cardiaco). In entrambi i casi l'alterazione del rapporto V/Q comporta il ritorno di sangue non ossigenato al cuore con conseguente ipossiemia. La risposta compensatoria all'ipossiemia ਠrappresentata dall'aumento della frequenza respiratoria con una conseguente maggiore eliminazione di CO2. ⢠Insufficienza respiratoria di tipo II: definita ipossiemico-ipercapnica. E' dovuta ad una condizione di ipoventilazione alveolare con conseguente incapacità del sistema respiratorio ad eliminare CO2 in modo adeguato. Si realizza pi๠frequentemente nelle condizioni che impediscono direttamente la ventilazione, quali: riduzione dell'input a livello del SNC (trauma cranico, emorragia intracranica, apnee della prematurità ) ; alterazioni delle giunzioni neuro-muscolari (danno al midollo spinale, avvelenamento da organofosfati/carbammati, sindrome di Guillain-Barrà¨, miastenia gravis, botulismo) e patologie neuromuscolari (miopatie e distrofie muscolari). Questa forma puಠinstaurarsi insidiosamente per il sopraggiungere della fatica dei muscoli respiratori come complicanza di una patologia preesistente (processo broncopneumonico acuto, stato di male asmatico, bronchiolite grave) esordita inizialmente con ipossiemia senza ipoventilazione. La sola supplementazione di ossigeno in questa forma di insufficienza respiratoria puಠnon essere appropriata. Questo ਠvero soprattutto in quelle condizioni cliniche nelle quali il soggetto si ਠadattato ad una condizione di ipercapnia cronica (come nei bambini con fibrosi cistica) ed ਠrelativamente dipendente dai chemocettori periferici ossigeno-sensibili per mantenere il drive ventilatorio. In questa forma il trattamento con solo ossigeno puಠportare ad una depressione del drive ventilatorio con aumento dei livelli di ipercapnia [1, 2]. L'insufficienza respiratoria in età pediatrica puಠessere inoltre classificata in acuta, cronica e cronica riacutizzata, in base al tempo intercorso tra la presentazione dei sintomi e il suo sviluppo. Nella forma acuta la compromissione della funzione respiratoria ਠspesso di entità grave e avviene in un periodo temporale molto breve (ore o giorni) ; nella forma cronica, invece, insorge lentamente (settimane o mesi) ed ਠdi severità minore per l'istaurarsi dei meccanismi di compenso; mentre la forma cronica riacutizzata rappresenta il deterioramento acuto di un'insufficienza respiratoria cronica [2]. Nella tabella 2 sono riportate le patologie nelle quali viene pi๠frequentemente utilizzata l'ossigenoterapia. L'inizio dell'ossigenoterapia ਠindicato per valori di PaO2 inferiori a 60 mmHg ed una SaO2 inferiore al 90%, e comunque in tutte quelle condizioni cliniche in cui ਠlegittimo sospettare una condizione di ipossia [4, 5, 6]. Quali segni clinici precoci di ipossia si possono considerare i seguenti: ⢠aumento della frequenza respiratoria e cardiaca in relazione all'età ; ⢠utilizzo dei muscoli respiratori accessori; ⢠ridotta tolleranza alla sforzo; ⢠irritabilità ; ⢠riduzione delle capacità mentali; ⢠insorgenza di crisi di apnea e bradicardia (soprattutto nei lattanti). Tra i segni pi๠tardivi si annoverano: ⢠stato confusionale; ⢠alterazioni dello stato di coscienza fino al coma; ⢠aritmie cardiache; ⢠cianosi [1, 7, 8].
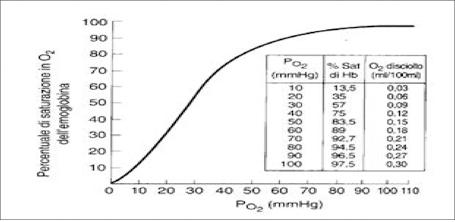
La pressione parziale di O2 (PaO2) nei soggetti normali ਠinfluenzata da numerosi fattori, principalmente l'età , l'altitudine e la frazione inspiratoria di ossigeno (FiO2). La relazione esistente tra PaO2 ed Hb viene rappresentata dalla curva di dissociazione dell'Hb (Figura 1). Per le caratterisiche proprie della curva si evince che a valori di PaO2 normali (>90 mmHg) l'Hb ਠsatura al 95% e la curva assume un andamento piatto. Di conseguenza un aumento di PaO2 (per iperventilazione o per somministrazione di ossigeno esogeno) comporterà solo un minimo e poco significativo incremento della concentrazione di ossigeno nel sangue. Al contrario, per valori <60 mmHg, ogni ulteriore caduta della PaO2 produce una variazione molto marcata della SaO2 (sO2 <90%) con evidenti ricadute sull'ossigenazione tessutale (Figura 1) [1, 2].
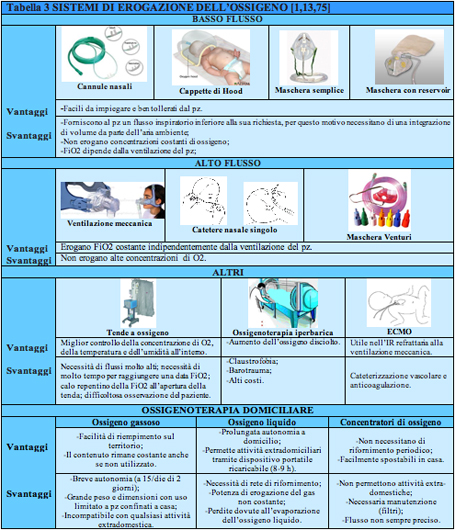
particolarmente utile nei casi di ipossiemia in presenza di una ventilazione sufficiente, in quanto permette una precisa valutazione della FiO2 ed ਠutile anche in caso di respirazione naso buccale o prevalentemente buccale. Le maschere con reservoir esistono in due varietà : la partial re-breathing mask e la non re-breathing mask. La prima ਠsprovvista di valvole unidirezionali tra maschera e reservoir, motivo per cui una parte dei gas espirati, circa un terzo, entra nel reservoir divenendo parte della successiva respirazione, mentre i restanti due terzi vengono allontanati attraverso apposite aperture nella maschera. Con tali maschere si raggiungono FiO2 di 0, 80. La seconda invece ਠdotata di una valvola unidirezionale tra maschera e reservoir, in modo che il bambino inali solo dal reservoir e possa espirare solo attraverso valvole ad una via poste sul bordo della maschera. Un sistema di valvole di sicurezza permette inoltre all'aria di entrare nel sistema nel caso in cui la sorgente di ossigeno venisse accidentalmente sconnessa. Con tale tipo di maschera sono raggiungibili FiO2 di 0, 95. Le indicazioni maggiori per questo tipo di presidio sono tutte le situazioni acute in cui vi sia la necessità di somministrare ossigeno ad alte concentrazioni e per un breve periodo. Le tende a ossigeno sono sistemi misti che possono usare tanto tecniche ad alto flusso quanto a basso flusso. Si tratta di dispositivi in materiale plastico che avvolgono completamente il letto del paziente (tabella 3). Consentono di controllare la concentrazione di ossigeno, la temperatura e l'umidità all'interno ed hanno il vantaggio, inoltre, di evitare al paziente il fastidio dell'applicazione di cannule, cateteri o maschere. Numerosi, tuttavia, sono gli inconvenienti legati al loro utilizzo, quali la necessità di flussi di ossigeno molto alti (10-15 l/min), la necessità di molto tempo per il raggiungimento di una data FiO2, il calo repentino della FiO2 all'apertura della tenda e la difficoltosa osservazione del paziente da parte del personale medico e dei genitori [1, 74]. Nei trattamenti a lungo termine ਠprevisto l'utilizzo del sistema a basso flusso, in grado di erogare anche quantità di 0, 1 l/min, in considerazione delle esigenze e dell'età del paziente. Le fonti attualmente disponibili per la somministrazione di ossigeno domiciliare sono, attualmente, le bombole ad alta pressione (gassoso), i sistemi ad ossigeno liquido e i concentratori di ossigeno (Tabella 3). La scelta dei diversi sistemi ਠlegata, oltre ai vantaggi e svantaggi, anche all'età del paziente, al livello di autonomia ed al flusso di ossigeno necessario. I dispositivi di erogazione dell'ossigeno comunemente utilizzati sono le cannule nasali, nei bambini con vie respiratorie integre, sostituite dalle maschere facciali nei soggetti con occlusione delle narici e/o che respirano a bocca aperta. Nei bambini tracheostomizzati, invece, la somministrazione di ossigeno attraverso la cannula tracheostomica, qualora ve ne sia precisa indicazione, puಠavvenire attraverso il collegamento mediante specifiche maschere per tracheotomia; tuttavia, essendo questo presidio difficilmente fissabile in un bambino, non consentendo quindi una somministrazione precisa dell'ossigenoterapia, si puಠovviare con l'uso del ânaso artificialeâ che consiste in un filtro umidificatore passivo che prevede una presa per l'ossigeno ed un foro centrale per l'aspirazione, che risulta di fatto, quello di uso pi๠comune, in quanto pi๠pratico ed efficace. Tuttavia, per tale presidio, ਠraccomandato l'uso su pazienti con peso corporeo superiore a 15 Kg. Per i bambini di peso corporeo inferiore non risulta ci siano attualmente strumenti certificati per la somministrazione di O2 domiciliare attraverso la tracheotomia [2, 76].
Durante la ventilazione meccanica, ਠpossibile migliorare l'ossigenazione aumentando la FiO2 o la pressione media delle vie aeree. La ventilazione meccanica viene iniziata per fornire un supporto a polmoni che funzionano normalmente o per malattie che fanno diminuire la compliance (sindrome da distress respiratorio acuto, atelettasia, polmonite, edema polmonare ed emorragia polmonare) o aumentare la resistenza (asma, bronchiolite, displasia broncopolmonare, inalazione di fumo e fibrosi cistica). Le situazioni in cui i polmoni sono normali possono non richiedere supplemento di ossigeno, in caso contrario si inizia con una FiO2 al 100% per poi ridurla al 50%. Le malattie di diminuita compliance, invece, causano una ipossiemia significativa ed ਠconsuetudine iniziare con una FiO2 al 100% per poi ridurla al 60% o meno al fine di evitare la tossicità da ossigeno. Nelle malattie di aumentata resistenza, infine, si inizia con una FiO2 al 100% riducendola lentamente al fine di mantenere un'adeguata ossigenazione ed evitando, al tempo stesso, una tossicità da ossigeno. Gli obiettivi principali della ventilazione meccanica sono: fornire un adeguato scambio di gas e favorire l'eliminazione dell'anidride carbonica senza causare un barotrauma polmonare o una tossicità da ossigeno. L'ECMO (ExtraCorporeal Membrane Oxygenation, ossigenazione di mambrana extracorporea) ਠusata nel trattamento di neonati e lattanti con insufficienza respiratoria refrattaria potenzialmente fatale che non risponde alla ventilazione meccanica e si prevede che si risolva in un breve periodo di tempo. Tuttavia a causa dei suoi rischi (da cateterizzazione vascolare e anticoagulazione) e del fatto che i suoi vantaggi rispetto al trattamento convenzionale nei pazienti non-neonatali non stati dimostrati in modo inequivocabile, le indicazioni per l'ECMO richiedono notevole esperienza, prudenza e giudizio [75, 77]. Bibliografia 1. Niccoli AA, Castellucci G. Insufficienza respiratoria e ossigenoterapia. Broncopneumologia. 2008;5-10. 2. Esposito F, Cavaliere P, Esposito I et al. Ossigenoterapia domiciliare nei bambini con insufficienza respiratoria cronica. Pneumologia Pediatrica 2009; 33: 31-42. 3. Wagstaff AT. Ossigenoterapia. In: Bersten A.D., Soni N. Oh Manuale di Terapia Intensiva. 6 th ed. Elsevier. 2010;319-31. 4. Kallstrom TJ. AARC Clinical Practice Guideline: oxygen therapy for adults in the acute care facility: 2002 revision & update. Respir Care 2002;47:717-20. 5. Duke T, Graham SM, Cherian MN. Oxygen is an essential medicine: a call for international action. Int J Tuberc Lung Dis. 2010;14 (11) :1362-8. 6. Ashworth A, Bickler S, Deen J. Guidelines for the management of common illnesses with limited resources. World Health Organization. 2005;72-5. 7. Philip Ayiekoa and Mike English. In Children Aged 2-59 months with Pneumonia, Which Clinical Signs Best Predict Hypoxaemia?. J Trop Pediatr. 2006;52:1-2. 8. Terzano C, Pacilio R. Ossigenoterapia. In: Terzano C. Malattie dell'apparato respiratorio. Springer. 2006:741-56. 9. Mac Lean JE, Fitzgerald DA. A rational approach to home oxygen use in infants and children. Pediatric Respiratory Reviews. 2006;7:215-22. 10. Statement on the care of the child with chronic lung disease of infancy and childood. Am J Respir Crit Care Med. 2003;168:356-96. 11. Katecha S, Allen J. Oxygen therapy for infants with chronic lung disease. Arch Dis Child Neonatal. 2002;87:11-4. 12. Indinnimeo L, Barbato A, Cutrera R et al. Gestione dell'attacco acuto di asma in età pediatrica: Linee Guida della Società Italiana di Pediatria. Pneumologia Pediatrica 2008;29:5-20. 13. Patria MF, GiannଠL, Colnaghi M et al. Il follow-up del neonato prematuro: quello che il pediatra deve conoscere. Pneumologia Pediatrica. 2005; 19: 13-23. 14. Marino PL, Conti G, Gattinoni L. The ICU book Terapia intensiva principi fondamentali. 3 th ed. Elsevier. 2007;389-404. 15. Chen M, àitil A, McCabe F. Infection, Oxygen, and Immaturity: Interacting Risk Factors for Retinopathy of Prematurity. Neonatology. 2011;99:125-132. 16. Sapieha P, Joyal JS, Rivera JC et al. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J Clin Invest. 2010;120 (9) :3022-32. 17. Figueras-Aloy J, Alvarez-Domànguez E, Morales-Ballus M et al. Early administration of erythropoietin in the extreme premature, a risk factor for retinopathy of prematurity?. An Pediatr. 2010;73 (6) :327-33. 18. Critical Care Monitoring Clinical Reference and Troubleshooting Guide of the Ge Medical Systems Information Technologies. 2005 General Electric Company. 19. Moon RE, Camporesi EM. Monitoraggio respiratorio. In: Miller RD, Fleisher LA, Johns RA et al. Anestesia. 6 th ed. Elsevier. 2006;l:1437-82. 20. Chierego ML, Nadalin S, Sallusti R et al. Medicina perioperatoria. In: Gullo A. Terapia intensiva Emergenza. Springer. 2003;3-130. 21. Urquhart DS, Montgomery H, Jaffe A. Assessment of hypoxia in children with cystic fibrosis. Arch Dis Child. 2005;90:1138-43. 22. De Martino G, D'Alicandro G, Vaira MI. Ossigenoterapia iperbarica. In: Mazzeo F. Trattato di clinica e terapia chirurgica. Piccin. 2001;1:999-1008. 23. Chenchen Wang, MD, MSc; Steven Schwaitzberg, MD; Elise Berliner. Hyperbaric Oxygen for Treating Wounds A Systematic Review of the Literature. Arch Surg. 2003;138:272-279. 24. G. Aprea, M. Brauzzi, C. Costanzo et al. Linee guida SIMSI: Medicina subacquea ed iperbarica. 2007:7-36. 25. Waisman D; Shupak A, Weisz G et al. Hyperbaric Oxygen Therapy in the Pediatric Patient: The Experience of the Israel Naval Medical Institute. Pediatrics. 1998;102 (5) :1-9. 26. Saugstad OD, Ramji S, Soll RF et al. Resuscitation of newborn infants with 21% or 100% oxygen: an updated systematic review and meta-analysis. Neonatology. 2008;94 (3) :176-82. 27. The International Liaison Committee on Resuscitation. The International Liaison Committee on Resuscitation (ILCOR) Consensus on Science With Treatment Recommendations for Pediatric and Neonatal Patients: Neonatal Resuscitation. Pediatrics 2006;117;978-88. 28. Thom SR. Ossigeno iperbarico in ICU. In: Fink MP, Abraham E, Vincent JL et al. Terapia intensiva. 5 th ed. Elsevier Masson. 2007;539-42. 29. Uzun G, Sen H, MutluoÄlu M et al. Hyperbaric oxygen therapy for pediatric patients with carbon monoxide poisoning. Turk J Pediatr. 2009;51 (4) :403-4. 30. Von Heimburg D, Noah EM, Sieckmann UP et al. Hyperbaric oxygen treatment in deep frostbite of both hands in a boy. Burns. 2001;27 (4) :404-8. 31. Wiebers J, Purdy I, Lieber M et al. Perinatal/Neonatal case presentation: Hyperbaric oxygen in treatment of neonatal arterial thromboembolism of lower extremities. Journal of Perinatology. 2006;26: 777-9. 32. Mazariegos GV, Toole KO, Mieles LA et al. Hyperbaric Oxygen Therapy for Hepatic Artery Thrombosis After Liver Transplantation in Children. Liver Transplantation and Surgery. 1999;5 (5) :429-36. 33. Chaudry T, McMahon A and Pilkington C. Wegener's Granulomatosis: paediatric presentation with ischaemia of the feet and novel use of hyperbaric oxygen. Pediatric Rheumatology. 2008;6 (1) :265. 34. Olivieri AN, Mellos A, Duilio C et al. Refractory vasculitic ulcer of the toe in adolescent suffering from Systemic Lupus Erythematosus treated successfully with hyperbaric oxygen therapy. Italian Journal of Pediatrics. 2010;36:72. 35. Alsheikheh A, Lieb W, Grehn F et al. Criswick-Schepens syndrome -- familial exudative vitreoretinopathy. Report of six cases in two consanguineous families. Ophthalmologe. 2004;101 (9) :914-8. 36. Minghua Chen a Ayse àitil a Frank McCabe b. Infection, Oxygen, and Immaturity: Interacting Risk Factors for Retinopathy of Prematurity. Neonatology. 2011;99:125-32. 37. Bernbeck B, Christaras A, Krauth K et al. Bone marrow oedema and aseptic osteonecrosis in children and adolescents with acute lymphoblastic leukaemia or non-Hodgkin-lymphoma treated with hyperbaric-oxygen-therapy (HBO) : an approach to cure? -- BME/AON and hyperbaric oxygen therapy as a treatment modality. Klin Padiatr. 2004;216 (6) :370-8. 38. Zhdanov GG, Nechaev VN, Alipov PA et al. Hyperbaric oxygenation and antioxidants in the complex intensive therapy of severe forms of pneumonia in children. Anesteziol Reanimatol. 1991; (2) :54-8. 39. Ponikvar R, ButuroviÄ J, Cizman M et al. Hyperbaric oxygenation, plasma exchange, and hemodialysis for treatment of acute liver failure in a 3-year-old child. Artif Organs. 1998 ;22 (11) :952-7. 40. Ohno Y, Kanematsu T. Hyperbaric oxygen therapy for intestinal obstruction in children: an exceptional experience in a compromised child. J Pediatr Surg. 1998;33 (10) :1543-5. 41. Maritschnegg P, Sovinz P, Lackner H et al. Granulomatous amebic encephalitis in a child with acute lymphoblastic leukemia successfully treated with multimodal antimicrobial therapy and hyperbaric oxygen. J Clin Microbiol. 2011;49 (1) :446-8. 42. Segal E, Menhusen MJ and Simmons S. Hyperbaric Oxygen in the Treatment of Invasive Fungal Infections: A Single-Center Experience. IMAJ 2007;9:355-7. 43. Vecchione R, Ruocco F, Colloca R et al. Pediatria e ossigenoterapia iperbarica: revisione della letteratura. Med Sub Iper. 1995;7-12. 44. Smith-Slatas CL, Bourque M and Salazar JC. Clostridium septicum Infections in Children: A Case Report and Review of theLiterature. Pediatrics. 2006;117:796-805. 45. Takac I, Kvolik S, Divkovic D et al. Conservative surgical management of necrotic tissues following meningococcal sepsis: case report of a child treated with hyperbaric oxygen. Undersea Hyperb Med. 2010;37 (2) :95-9. 46. Siraneci R, HatipoÄlu N, HatipoÄlu H et al. Acute arterial thrombotic purpura complicating varicella and the role of hyperbaric oxygen as an adjunctive therapy. Turk J Pediatr. 2004;46 (3) :256-8. 47. Krzelj V, Petri NM, Mestrovic J et al. Purpura fulminans successfully treated with hyperbaric oxygen--a report of 2 cases. Pediatr Emerg Care. 2005;21 (1) :31-4. 48. Hsieh WS, Yang PH, Chao HC et al. Neonatal Necrotizing Fasciitis: A Report of Three Cases and Review of the Literature. Pediatrics. 1999;103;53. 49. Murphy JJ, Granger R, Blair GK et al. Necrotizing fasciitis in childhood. J Pediatr Surg. 1995 Aug;30 (8) :1131-4. 50. Kurschel S, Mohia A, Weigl V et al. Hyperbaric oxygen therapy for the treatment of brain abscess in children. Childs Nerv Syst. 2006;22 (1) :38-42. 51. Baechli H, Schmutz J, Mayr JM et al. Hyperbaric oxygen therapy (HBO) for the treatment of an epidural abscess in the posterior fossa in an 8-month-old infant. Pediatr Neurosurg. 2008;44 (3) :239-42. 52. Saunders PJ. Hyperbaric oxygen therapy in the management of carbon monoxide poisoning, osteoradionecrosis, burns, skin grafts, and crush injury. Int J Technol Assess Health Care. 2003 Summer;19 (3) :521-5. 53. Stoller KP. Quantification of Neurocognitive Changes Before, During, and After Hyperbaric Oxygen Therapy in a Case of Fetal Alcohol Syndrome. Pediatrics. 2005;116;586-591. 54. Collet JP, Vanasse M, Marois P et al. Hyperbaric oxygen for children with cerebral palsy: a randomised multicentre trial. Lancet. 2001;357 (9256) :582-6. 55. Papazian O, Alfonso I. Hyperbaric oxygen treatment for children with cerebral palsy. Rev Neurol. 2003;37 (4) :359-64. 56. Muller-Bolla M, Collet JP, Ducruet T et al. Side effects of hyperbaric oxygen therapy in children with cerebral palsy. Undersea Hyperb Med. 2006;33 (4) :237-44. 57. McDonagh MS, Morgan D, Carson S et al. Systematic review of hyperbaric oxygen therapy for cerebral palsy: the state of the evidence. Developmental Medicine & Child Neurology. 2007;49:942-7. 58. Zhou BY, Lu GJ, Huang YQ et al. Efficacy of hyperbaric oxygen therapy under different pressures on neonatal hypoxic-ischemic encephalopathy. Zhongguo Dang Dai Er Ke Za Zhi. 2008;10 (2) :133-5. 59. Rossignol DA, Rossignol1 LW, James SJ et al. The effects of hyperbaric oxygen therapy on oxidative stress, inflammation, and symptoms in children with autism: an open-label pilot study. BMC Pediatrics. 2007;7:36. 60. Rossignolab DA, Rossigno LW. Hyperbaric oxygen therapy may improve symptoms in autistic children. Med Hypotheses. 2006;67 (2) :216-28. 61. Buompadre MC, Arroyo HA. Accidental cerebral venous gas embolism in a young patient with congenital heart disease. J Child Neurol. 2008;23 (1) :121-3. 62. French LK, Horowitz BZ, McKeown NJ. Hydrogen peroxide ingestion associated with portal venous gas and treatment with hyperbaric oxygen: a case series and review of the literature. Clin Toxicol. 2010;48 (6) :533-8. 63. Beltz K, Christaras A, Kovacevic A et al. A novel element in the management of chronic granulomatous disease (CGD) ? - treatment of osteomyelitis with additional hyperbaric oxygen therapy (HBO). Klin Padiatr. 2008;220 (6) :380-3. 64. Paim LB, Liphaus BL, Rocha AC et al. Chronic recurrent multifocal osteomyelitis of the mandible: report of three cases. J Pediat. 2003;79 (5) :467-70. 65. Chuba PJ, Aronin P, Bhambhani K. Hyperbaric Oxygen Therapy for Radiation-Induced Brain Injury in Children. Cancer. 1997;80 (10) :2005-12. 66. Stankovà¡ J, Kavan P, Kràzovà¡ H et al. 131I meta-iodobenzylguanidine in combination with hyperbaric oxygen therapy in the treatment of prognostically high-risk forms of neuroblastoma. Cas Lek Cesk. 2001;140 (1) :13-7. 67. Asharnalla HL, Goldwein JW, Thorn SR et al. Hyperbaric Oxygen Therapy for the Treatment of Radiation-Induced Sequelae in Children The University of Pennsylvania Experience. Cancer. 1996;77 (11) :2407-12. 68. Rapley JH, Lawrence WT, Witt PD et al. Composite grafting and hyperbaric oxygen therapy in pediatric nasal tip reconstruction after avulsive dog-bite injury. Ann Plast Surg. 2001;46 (4) :434-8. 69. McCrary BF. Hyperbaric oxygen (HBO2) treatment for a failing facial flap. Postgrad Med J. 2007;83. 70. Bennett M, Heard R. Hyperbaric oxygen therapy for multiple sclerosis. CNS Neurosci Ther. 2010;16 (2) :115-24. 71. Bennett M, Heard R. Hyperbaric oxygen therapy for multiple sclerosis. Cochrane Database Syst Rev. 2004; (1). 72. OlszaÅski R, Pachut M, SiÄko Z et al. Efficacy of hyperbaric oxygenation in atopic dermatitis. Bull Inst Marit Trop Med Gdynia. 1992;43 (1-4) :79-82. 73. Battistini E, Fasce L, Defilippi AC et al. Asma e near fatal asthma. In: Lorini R, Di Pietro P, Romano C et al. Pediatria d'urgenza. Masson. 2005;431-43. 74. Sà nchez Sà nchez MM. Ossigenoterapia. In: Moreno ML, Rivera SA, de la Torre AE. Il paziente critico. Protocolli e procedure di assistenza generale e specialistica. Masson. 2005; 550-5. 75. Frankel LR. Distress respiratorio e insufficienza respiratoria. In: Kliegman RM, Behrman RE, Jenson HB et al. Nelson Textbook of Pediatrics. 18th ed. Elsevier. 2009;438-41. 76. Balfour-Lynn IM, Field DJ, Gringras P et al. BTS guidelines for home oxygen in children. Thorax. 2009;64 (2) :1-26. 77. Lorry R, Kache F, Kache S. Ventilazione meccanica. In: Kliegman RM, Behrman RE, Jenson HB et al. Nelson Textbook of Pediatrics. 18th ed. Elsevier. 2009;18:442-8.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Obesità infantile: natura o cultura?
Valeria Chirico, Rosangela Caruso, Carbone Maurizio, Caterina MunafಠDipartimento di Scienze Pediatriche Mediche e Chirugiche, Università di Messina
L'obesità ਠuna malattia complessa dovuta a fattori genetici, ambientali ed individuali con conseguente alterazione del bilancio energetico ed accumulo eccessivo di tessuto adiposo nell'organismo. L'obesità ਠsempre pi๠oggetto di attenzione da parte del mondo medico in quanto la sua progressiva diffusione preoccupa, tanto da essere stata definita dall'Organizzazione Mondiale della Sanità (OMS) un' âepidemia globaleâ. I dati forniti dall'ISTAT documentano che in Italia nella fascia di età degli 8 anni il 36% dei bambini ha problemi di eccesso di peso, di cui il 24% ਠin sovrappeso e il 12% ਠdecisamente obeso. Complessivamente ਠsempre il Meridione a presentare prevalenze di obesità e sovrappeso pi๠elevate ma, per la prima volta, si osserva un aumento del tasso di obesità nel Nord-Ovest, area tradizionalmente a pi๠bassa prevalenza, che supera quello del Centro.
Sono stati identificati quattro periodi critici per lo sviluppo dell'obesità : Vita intrauterina : Viene plasmato l'assetto metabolico del feto in relazione a quello della madre. L'esposizione del feto a ipernutrizione durante la gravidanza puಠcontribuire alla âprogrammazioneâdella regolazione metabolica del bambino nella sua vita adulta, predisponendo l'individuo all'obesità e al diabete. Allo stesso modo anche l'esposizione all'iponutrizione intrauterina ਠstata associata ad una pi๠elevata prevalenza dell'obesità nei giovani adulti. Primo anno di vita: L'elevata velocità di crescita ponderale e staturale durante il primo anno di vita, favorita da un grande apporto in calorie e nutrienti, ਠassociata al sovrappeso ed all'obesità nelle età successive. L'allattamento al seno soprattutto se prolungato per pi๠di sei mesi, ਠun fattore protettivo nei confronti dell'obesità Età prescolare: adiposity rebound: Si intende l'inversione della curva dell'adiposità che avviene fisiologicamente al sesto-settimo anno di vita. I bambini che presentano un'anticipazione dell'adiposity rebound hanno un elevato rischio di diventare obesi nelle età successive. Pubertà : In questa fase vi ਠil rischio che la deposizione di lipidi sia eccessiva, in relazione alla riduzione di attività fisica e sportiva, soprattutto nelle femmine, e al generale peggioramento delle abitudini nutrizionali tipiche dell'adolescenza, soprattutto lo snacking fuoripasto.
Le scelte nutrizionali: Tra i diversi aspetti dell'alimentazione si sta cercando di capire quali contribuiscano in modo particolare all'eccesso ponderale. Una dieta ad alto carico glicemico rappresenta un fattore di rischio per lo sviluppo di adiposità centrale, cardiovasculopatie e diabete mellito. Gli errori alimentari maggiormente diffusi nei bambini obesi si possono cosଠriassumere: - eccessiva assunzione di cibo rispetto al consumo calorico (vita troppo sedentaria) - mancata assunzione della prima colazione - i bambini obesi che effettuano la prima colazione tendono comunque a mangiare poco al mattino - tendenza ad assumere alimenti preferibilmente nel pomeriggio e alla sera, spesso non in occasione dei pasti principali - scarso apporto di: cereali, legumi, pesce, fibra alimentare, verdura e frutta di stagione - elevato apporto di lipidi: salumi, formaggi e carne, dolci - elevato apporto di zuccheri ad alto indice glicemico (patate, pane, cereali raffinati) - preferenza per i cibi liquidi (per esempio succhi di frutta) - due condotte alimentari sembrano, in particolare, specifiche del bambino e dell'adolescente obeso: l'iperfagia e il piluccamento, spesso a connotazione familiare.
Un'iperalimentazione nei primi due anni di vita oltre a causare un aumento di volume delle cellule adipose (ipertrofia), determina anche un aumento del loro numero (iperplasia) . Numerosi sono gli studi che hanno dimostrato un'associazione positiva tra adiposità e âskipping breakfastâ. Esistono studi che dimostrano come una ricca colazione, sia in bambini normopeso che obesi, abbia un impatto significativo sull'assunzione di alimenti a pranzo riuscendo a ridurre gli eccessi nel pasto successivo. La validità della prima colazione puಠessere, quindi, individuata nella sua capacità di regolare l'assunzione calorica nei pasti successivi e nell'integrazione di alcuni micronutrienti (per esempio ferro e zinco) che altrimenti sono spesso carenti, specie nelle adolescenti. Una recente indagine longitudinale di Berkey ha studiato la frequenza di assunzione del breakfast, l'alimentazione e il BMI in una popolazione di 14 mila ragazzi tra i 9 e i 14 anni. A conferma di altri studi, si rileva che i bambini che saltano la prima colazione hanno una dieta meno salutare, caratterizzata da una maggior quota di lipidi e un pi๠basso intake di vitamine e minerali. I bambini che non mangiano mai la prima colazione sono di peso superiore ai coetanei che invece hanno l'abitudine di assumerla. Negli ultimi decenni si ਠassistito, insieme ad un generale incremento degli apporti in zuccheri a rapido assorbimento nell'alimentazione della popolazione in tutto il mondo, anche ad un progressivo aumento del consumo di bevande zuccherate. Un gruppo di 548 adolescenti ਠstato seguito per un anno e mezzo con frequenti valutazioni antropometriche e degli apporti nutrizionali. In questi ragazzi, il consumo di bevande zuccherate ਠrisultato associato all'incremento dell'adiposità nel corso dello studio. In particolare, per ogni porzione al dଠdi bevanda zuccherata consumata aumentava, dopo aggiustamento per caratteristiche antropometriche e demografiche, variabili nutrizionali e stile di vita, sia il BMI (0.24 kg/m2/porzione/die) al momento del reclutamento, che lâincremento del BMI (0.18 kg/m2/porzione/die) nel corso del follow-up. Inoltre, uno studio osservazionale prospettico ha suggerito che il rischio di sviluppo di obesità aumenta del 60 per cento nei bambini della scuola media per ogni bibita al giorno in pià¹, anche dopo la correzione per eventuali fattori confondenti. Le bibite zuccherate promuovono l'apporto energetico e l'eccessivo aumento ponderale a causa della loro influenza sul metabolismo glucidico. Per contro, il latte e lo yogurt, alimenti a basso indice glicemico, sembrano proteggere gli adolescenti in sovrappeso dal rischio di diventare obesi. Un' indagine mirata ai fuoripasto e condotta su di un campione di 1800 bambini di età compresa tra 7 e gli 11 anni reclutati nell'Italia settentrionale, centrale e meridionale, ha evidenziato un consumo medio di fuoripasto pari a 3 porzioni al giorno. I fuoripasto pi๠consumati, con apporti medi di una porzione al giorno, sono risultati le bevande zuccherate (nell'ordine succhi di frutta, the, bibite gassate) . I due rimanenti fuoripasto giornalieri sono costituiti in primis dal panino imbottito, seguito da crakers, schiacciate e frutta.
Le abitudini motorie: Oltre all'aumentato consumo di cibi ricchi di energia e ricchi di grassi, la generale tendenza alla sedentarietà ha contribuito in modo notevole al vistoso incremento della prevalenza dell'obesità . La riduzione dell'attività fisica quotidiana, causa importante dello sviluppo del sovrappeso, ਠprogressiva con l'aumentare dell'età soprattutto nelle ragazze. Esiste una relazione inversamente proporzionale tra il peso corporeo e le ore dedicate all'attività fisica la quale riveste un ruolo protettivo nei confronti dell'eccessivo incremento di peso anche nell'età infantile. L'esercizio fisico ਠdi fondamentale importanza per il bambino che cresce, in quanto, oltre a farlo dimagrire, lo rende pi๠attivo, contribuendo a ridistribuire le proporzioni tra massa magra (tessuto muscolare) e massa grassa (tessuto adiposo) . Nell'ambito delle attività sedentarie, che occupano sempre maggior spazio nella giornata dei ragazzi, televisione e computer occupano molte ore con effetti importanti: riduzione del metabolismo, visione di pubblicità alimentari, invito a mangiare, sottrazione di tempo ad attività pi๠dispendiose. Le stime pi๠recenti indicano che i bambini necessitano di un minimo di sessanta minuti di attività fisica moderata-vigorosa al giorno.
Lo stile di vita: Le persone che soffrono di insonnia, e che non dormono rispettando le canoniche 8 ore di sonno, tendono a mangiare di pi๠rischiando l'obesità . Il tutto ਠriconducibile all'azione di due ormoni chiave atti alla regolazione dell'appetito: la grelina e la leptina. La grelina aumenta le sensibilità alla fame mentre la leptina tiene sotto controllo l'appetito. Chi dorme regolarmente per cinque ore a notte ha in media il 15% di grelina in pi๠e il 15% di leptina in meno rispetto a chi dorme per otto ore. Si ਠosservato che poche ore di sonno portano a una riduzione dei livelli di leptina ed a pi๠elevati livelli di grelina. Ovviamente lo stimolo della fame generato dalla grelina ha maggiore incidenza sul rischio di obesità .
Le occupazioni durante il tempo libero: à stata pi๠volte sottolineata la correlazione tra le ore trascorse davanti alla televisione ed il grado di sovrappeso, logica conseguenza di uno squilibrio tra introito e dispendio energetico. Vari studi hanno riscontrato una relazione direttamente proporzionale tra le ore trascorse davanti alla televisione e la prevalenza dell'obesità tra i bambini in età scolare. Una recente meta-analisi ha definitivamente confermato l'associazione tra esposizione video e obesità nei bambini già da tempo riportata anche nel nostro paese . Il video, in particolare la TV, ਠun fattore di rischio sia per la sedentarietà che per il consumo di cibo.
Marshall SJ, Biddle SJ, Gorely T, Cameron N, Murdey I. Relationships between media use, body fatness and physical activity in children and youth: a meta-analysis.Int J Obes Relat Metab Disord. 2004;28:1238-46.
Epstein LH, Roemmich JN, Paluch RA, Raynor HA. Influence of changes in sedentary behavior on energy and macronutrient intake in youth. Am J Clin Nutr. 2005;81:361-6.
Caroli M, Argentieri L, Cardone M, Masi A. Role of television in childhood obesity prevention. Int J Obes Relat Metab Disord. 2004; Suppl 3:S104-8.
Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet. 2001;357:505-8.
Snyder EE, Walts B, Perusse L, Chagnon YC, Weisnagel SJ, Rankinen T, Bouchard C. The human obesity gene map: the 2003 update. Obes Res. 2004;12:369-439.
Agostoni C. Breast-feeding and Childhood Obesity. Pediatr Res 2000; 47:3.
Tin SP, Ho SY, Mak KH, Wan KL, Lam TH. Breakfast skipping and change in body mass index in young children.Int J Obes. 2011 Mar 29.
Maffeis C, Surano MG, Cordioli S, Gasperotti S, Corradi M, Pinelli L. A high-fat vs. a moderate-fat meal in obese boys: nutrient balance, appetite, and gastrointestinal hormone changes. Obesity2010 ;18:449-55.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno III numero 2 - aprile 2011 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
L'aerosolterapia
INTRODUZIONE La terapia inalatoria ha origini antiche, quando all'acqua in ebollizione erano aggiunti principi attivi o presunti tali e successivamente inalati. 1 Il primo sistema per aerosol dell'era moderna fu ideato dal Dr. John Mudge nel 1778 (Figura 1). Da allora la terapia aerosolica ha subito una notevole diffusione non solo nell'età adulta ma anche in quella pediatrica e assunto validità scientifica e terapeutica innegabili per il trattamento di numerose patologie. 2, 3 Oggi sono disponibili una vasta gamma di apparecchi (devices) che amplificano notevolmente le possibilità di scelta, ma creano allo stesso tempo dubbi e incertezze sia tra i pazienti che tra i medici. 4 Diversi studi hanno, infatti, messo in luce l'inadeguata conoscenza da parte del personale medico riguardo l'utilizzo dei devices .5-9 Risulta quindi necessaria una rivoluzione culturale che partendo dai medici porti ad una maggiore consapevolezza e competenza dei pazienti.
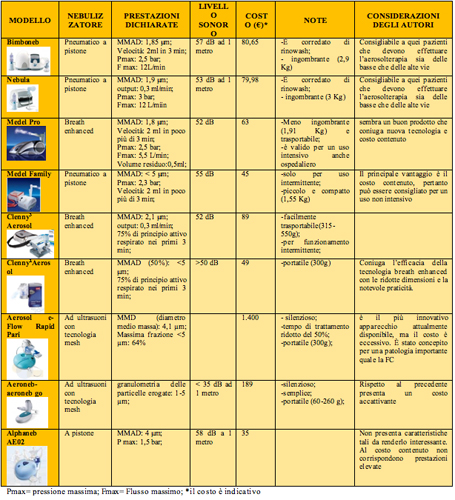
Confrontando i nebulizzatori classici, quelli âmiglioratiâ e quelli âattivatiâ dal respiro, questi ultimi due sono risultati superiori. In particolare, i nebulizzatori breath-enhanced hanno ridotto il tempo di trattamento, i breath-actuated hanno ridotto lo spreco di farmaco e incrementato la quota di farmaco inalata. 30, 33 Pi๠recenti sono i âmesh nebulizersâ (Figura 4) aventi come elemento pi๠importante e innovativo una membrana metallica micro perforata attraverso una tecnologia laser.
Figura 4 - esempi di "mesh nebulizers" commercializzati in Italia Un attuatore piezoelettrico, controllato da un circuito elettronico, mette in vibrazione la membrana, creando una differenza di pressione fra la camera contenente il farmaco e il nebulizzatore vero e proprio. Il farmaco allo stato liquido tende a spostarsi verso l'ambiente a pressione minore attraversando i micro fori della membrana e trasformandosi in aerosol, le cui particelle hanno dimensioni determinate dalla sezione dei micro fori stessi (Figura 5). 31
Figura 5 - Principio di funzionamento dei "mesh nebulizers" La tecnologia mesh ha dimostrato in diverse condizioni, sia in adulti che in bambini, di produrre una quantità di aerosol maggiore rispetto ai nebulizzatori tradizionali, in conseguenza della particolare modalità di produzione delle particelle respirabili, della percentuale di dose emessa e del residuo di farmaco rimanente nell'apparecchio. 34 I vantaggi offerti sono molteplici sia in termini di efficacia (basti pensare che le dimensioni delle particelle da inalare sono perfettamente predeterminate, una maggiore quota di farmaco raggiunge le basse vie aeree, con riduzione della deposizione orofaringea e degli effetti collaterali), sia in termini di compliance (portabilità , assenza di rumore, facilità d'uso). I limiti di questi apparecchi sono il costo non indifferente e il fatto che non possono nebulizzare farmaci viscosi. 31, 35 La tabella 1 mette a confronto alcuni dei nebulizzatori attualmente in commercio in Italia. UNO SGUARDO AL FUTURO: I-neb AAD System Nuovi nebulizzatori, definiti âintelligentiâ stanno per arrivare (Figura 6). 36 Essi combinano la tecnologia della membrana forata vibrante con quella AAD (Adaptive Aerosol Delivery).
Figura 6 - Il nuovo Respironics I-neb con tecnologia AAD eroga l'aerosol all'inizio dell'inspirazione Presentano numerosi vantaggi: minore volume residuo del farmaco, maggiore precisione della dose erogata in quanto si adattano al respiro del paziente senza spreco di farmaco durante l'espirazione, maggiore aderenza alla terapia grazie alla presenza di un segnale visivo, sonoro o vibratorio che informa il paziente della corretta e completa esecuzione della manovra. Si tratta di apparecchi silenziosi, portatili e ricaricabili, che consentono, tramite una singola piattaforma, la somministrazione di pi๠farmaci. La novità pi๠sorprendente e rivoluzionaria ਠla presenza di un chip di memoria che registra e trasmette i dati tramite internet ad un computer consentendo al medico di poter esaminare e monitorare in tempo reale la performance del paziente. Tramite questo sistema sono possibili anche la prescrizione e l'ordinazione elettronica del farmaco che verrà spedito a casa dalla farmacia. I primi studi hanno evidenziato il possibile utilizzo di I- neb AAD System per la somministrazione di α1-antitripsina in pazienti con Fibrosi Cistica (FC).36-39 Un altro studio ha testato questo apparecchio proprio su di un gruppo di pazienti con FC, con un evidente risparmio di tempo ed elevati livelli di compliance. 40 Recentemente, questo dispositivo ਠstato introdotto negli Stati Uniti per la somministrazione di iloprost nel trattamento dell'ipertensione polmonare, ma se risulterà realmente efficace la sua applicazione potrebbe estendersi al trattamento di tutte le patologie respiratorie. 36, 37
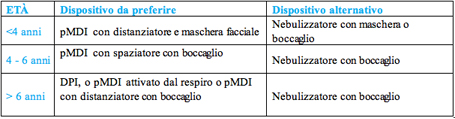
Tabella 3 Primo parametro da prendere in considerazione ਠl'età del paziente. Le Linee Guida SIP, facendo riferimento alle GINA 2006 (recentemente aggiornate), indicano chiaramente che fino ai 6 anni il dispositivo di prima scelta ਠil pMDI con distanziatore, mentre per i bambini che hanno pi๠di 6 anni gli spray predosati sono equivalenti agli erogatori di polvere. In tutte le età il nebulizzatore viene indicato come dispositivo di seconda scelta (tabella 4). 21, 84
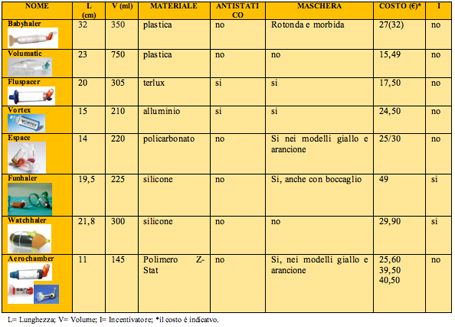
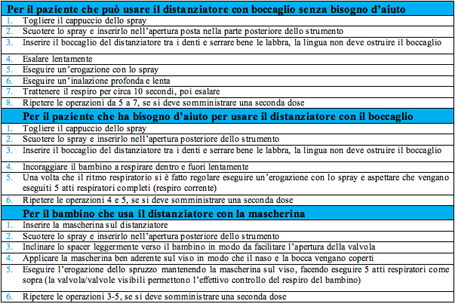
Tabella 5 - Le tecniche inalatorie consigliate92 LE CARICHE ELETTROSTATICHE Quando si usa un distanziatore costruito con un materiale non conduttore, come la plastica, la presenza di cariche elettrostatiche puಠridurre la quantità di farmaco inalata e rendere di volta in volta variabile la dose somministrata. Infatti, parte del medicinale spruzzato nello spacer aderisce alle pareti e diventa indisponibile per l'inalazione. Alcuni dei nuovi distanziatori hanno limitato questo problema, per esempio NebuChamber, Vortex, Fluspacer, OptiChamber e AeroChamber Plus.2, 97 In alternativa ਠpossibile ridurre le cariche elettrostatiche con un'appropriata pulizia immergendo lo spacer in acqua con poche gocce di un detergente ionico (il comune detersivo per lavare i piatti), lasciandolo asciugare da solo, senza strofinarlo con il panno, per 12-24 ore e ripetendo tale procedura una volta a settimana (tabella 6).71, 92, 97
Tabella 7 Istruire il paziente non ਠfacile ma ਠmeglio che cambiare semplicemente il device alla prima difficoltà .4, 22 Tutti i devices richiedono una qualche spiegazione e prescrivere un device al paziente non assicura un suo corretto uso.2 à necessario un nostro cambiamento per essere sicuri che i pazienti effettuino l'aerosol terapia con appropriatezza ed efficacia ed ਠfondamentale far capire l'importanza di una terapia fatta correttamente in primis ai genitori dei nostri piccoli.22, 113 Diversi studi hanno dimostrato le scarse conoscenze teoriche e pratiche di medici, infermieri e specializzandi inerenti l'uso dello spray con il distanziatore. à importante colmare queste gravi lacune perchਠsolo un medico con corrette abilità teorico-pratiche puಠimpartire un efficace insegnamento ai propri pazienti.5-9, 92, 115 Bibliografia 1. Milanesi E, Romei I, Piacentini G. Aerosolterapia delle basse vie aeree: i presupposti fisiopatologici. PneumologiaPediatrica2003; 12: 23-7. 2. Rubin BK. Air and Soul: The Science and Application of Aerosol Therapy. RESPIRATORY CARE. July 2010; 55 (7) : 911-21. 3. Carboni G, Carta G, Corona GB et al. Aerosolterapia: le indicazioni cliniche. Pneumologia Pediatrica. 2003; 12: 57-62. 4.Chrystyn H, Price D. Not all asthma inhalers are the same: factors to consider when prescribing an inhaler. Primary Care Respiratory Journal (2009) ; 18 (4) : 243-9. 5. Guidry CG, Brown WD, Stogner SW, et al. Incorrect use of metered dose inhalers by medical personnel. Chest 1992; 101:31-3. 6. Hanania NA, Wittman R, Kesten S, et al. Medical personnel's knowledge of and ability to use inhaling devices: metered-dose inhalers, spacing chambers, and breath- actuated dry powder inhalers. Chest 1994; 105:111-6. 7. Kesten S, Zive K, Chapman KR. Pharmacist knowledge and ability to use inhaled medication delivery systems. Chest 1993; 104:1737-42. 8. Lindgren S, Bake B, Larsson S. Clinical consequences of inadequate inhalation technique in asthma therapy. Eur J Respir Dis 1987; 70:93-8. 9. McFadden ER Jr. Improper patient techniques with metered dose inhalers: clinical consequences and solutions to misuse. J Allergy Clin Immunol 1995; 96:278-82. 10. Terzano C, Mannino F. Aerosol. Caratteristiche, analisi, applicazioni terapeutiche. McGraw Hill Ed 1997. 11. Corsico R. Tecniche di aerosol terapia. In: âtrattato Italiano di Allergologiaâ. Volume 2. Pavia: Selecta Medica ED. 2002; 1115-25. 12. Tiddens HA. Facts and fiction in inhalation therapy. Ital J Pediatr. 2003; 29: 39-43. 13. Dolovich MB, Ahrens RC, Hess DR et al. Device Selection and Outcomes of Aerosol Therapy: Evidence-Based Guidelines. Chest 2005;127;335-71. 14. Pedersen S, Dubus JC, Crompton G, on behalf of the ADMIT Working Group. The ADMIT series - Issues in Inhalation Therapy. 5) Inhaler selection in children with asthma. Primary Care Respiratory Journal (2010) ; 19 (3) : 209-16. 15. Gelardi M, Mappa L, Fiorella ML, Cassano M. Gli strumenti. Pneumologia Pediatrica 2003; 12: 8-14. 16. Rau JL. The inhalation of drugs: advantages and problems. Respir Care 2005;200:50 (8) :367-382. 17. Rubin BK, Kelly HW. The impact of drug delivery devices in asthma management. J Respir Dis 2002;4 (Suppl) :S36-S43. 18. Indinnimeo L, de Castro G, Tancredi G et al. Aerosolterapia delle alte vie aeree: le indicazioni cliniche. Pneumologia Pediatrica. 2003; 12: 15-22. 19. Varricchio A, De Lucia A, Tricarico D. Tecniche di terapia inalatoria nelle patologie delle vie aeree superiori (V.A.S.). Pneumorama 36/X/3-2004.23-7. 20. La Rosa Mario, Miraglia Del Giudice M. La terapia inalatoria in pediatria. Pneumologia Pediatrica. 2003; 12: 28-35. 21. Global Initiative for Asthma. Global strategy for asthma management and prevention 2009. www.ginasthma.com 22. Melani AS. Inhalatory therapy training: a priority challenge for the Physician. ACTA BIOMED 2007; 78: 233-45. 23. Lowenthal D, Kattan M. Facemasks versus mouthpieces for aerosol treatment of asthmatic children. Pediatr Pulmonol 1992;14 (3) :192-6. 24. Neuman SP, Clark AR, Talee N, Clarke SW. Lung deposition of 5 mg Intal from a pressurizated metered dose inhaler assessed by radiotracer technique. Int J Pharm 1991; 74: 203-208. 25. BerlinskiA, WaldrepJC. Nebulized drug admixtures: effect on aerosol characteristics and albuterol output. J Aerosol Med 2006; 19: 484-90. 26. Melani AS, Sestini P, Aiolfi S, Barbato N, Canessa P, De Angelis G, Zanchetta D, De Tullio R, Cinti C, Neri M, on be half of the Associazione Italiana Pneumologi Ospedalieri (AIPO) Educational Group. Home nebulizer use and maintenance in Italy. Eur Respir J 2001; 18: 758-63. 27. Hess D, Fisher D, Williams P et al. Medication Nebulizer Performance: Effects Of Diluent Volume, Nebulizer Flow, and Nebulizer Brand. Chest 1996;110;498-505. 28. Rubin BK. Nebulizer therapy for children: the device-patient interface. Respir Care 2002;47 (11) :1314-9. 29. ERS Task Force: European Respiratory Society Guidelines on the use of nebulizers. ERJ 2001; 18: 228-42. 30. Leung K, LoucaE, Coates AL. Comparison of Breath-Enhanced to Breath-Actuated Nebulizers for Rate, Consistency, and Efficiency. Chest 2004;126;1619-27. 31. Hess DR. Aerosol Delivery Devices in the Treatment of Asthma. RESPIRATORY CARE 2008;53 (6) :699 -723. 32. Hess DR, Myers TR, Rau JL with a Foreword by Sam Giordano, Executive DirectorAmerican Association for Respiratory Care. A Guide to Aerosol Delivery Devices For Respiratory Therapists. 2007. 33. Rau JL, Ari A and Restrepo RD. Performance Comparison of Nebulizer Designs: Constant-Output, Breath-Enhanced, and Dosimetric. Respir Care 2004;49 (2) :174-9. 34. Ari A, Atalay OT, Harwood R et al. Influence of Nebulizer Type, Position, and Bias Flow onAerosol Drug Delivery in Simulated Pediatric and Adult Lung ModelsDuring Mechanical Ventilation. Respir Care 2010;55 (7) :845-51. 35.Lass JS, Sant A, Knoch M. New advances in aerosolised drug delivery: vibrating membrane nebuliser technology. Expert opin Drug Deliv 2006 Sep;3 (5) :693-702. 36. Dhand R. Intelligent Nebulizers in the Age of the Internet:The I-neb Adaptive Aerosol Delivery (AAD) System. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY.2010; 23 (1) : iii-v. 37. Denyer J, Dyche T. The Adaptive Aerosol Delivery (AAD) Technology:Past, Present, and Future. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY. 2010; 23 (1) :S1-S10. 38. Denyer J, Prince I, Dixon E et al. Evaluation of the Target Inhalation Mode (TIM) Breathing Maneuver in Simulated Nebulizer Therapyin Patients with Cystic Fibrosis. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY. 2010; 23 (1) : S29-S36. 39. Geller DE, Kesser KC. The I-neb Adaptive Aerosol Delivery System EnhancesDelivery of a1-Antitrypsin with Controlled Inhalation. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY. 2010; 23 (1) : S55-S59. 40. Denyer J, Black A, Nikander K et al. Domiciliary Experience of the Target Inhalation Mode (TIM) Breathing Maneuver in Patients with Cystic Fibrosis. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY. 2010; 23 (1) : S45-54. 41. Pedersen S, Hansen R, Fuglsang G. Influence of inspiratory flow rate upon the effect of a Turbuhaler. Archives of Disease in Childhood 1990; 65: 308-19. 42. Chrystyn H. Is inhalation rate important for dry powder inhalers? Using the In-Check Dial to identify these rates. Resp Med 2003;97:181-7. 43. Everard ML, Devadason SG, Le Souef PN. Flow early in the inspiratory manoeuvre affects the aerosol particle size distribution from a Turbuhaler. Respir Med 1997;91 (10) :624-8. 44. Svartengren K, Lindestad P, Svartengren M, Philipson, Bylin G and Camner P. Added external resistance reduces oropharyngeal deposition and increases lung deposition of aerosol particles in asthmatics. Am J RespirCrit Care Med 1995;152:32-7. 45. Brindley A, Sumby BS, Smith IJ, Prime D, Haywood PA, Grant AC. Design, manufacture and dose consistency of the Serevent Diskus inhaler. Pharm Technol Eur 1995; 7: 14-22. 46. WetterlinK. Turbuhaler: a new powder inhaler for administration of drugs to the airways. Pharm Res 1988; 5: 506-8. 47. Fuller R. The Diskus: a new multi-dose powder device. Efficacy and comparison with Turbuhaler. J Aerosol Med 1995; 8: S11-S17. 48. Chrystyn H. The DiskusTM: a review of its position among dry powder inhaler devices. Int J ClinPract. June 2007, 61, 6, 1022-36. 49. Battistini E. Erogatori di polvere. Pneumologia Pediatrica 2003; 12: 49-56. 50. Broeders ME, Molema J, Vermue NA, Folgering HTM. Peak inspiratory flow rate and slope of the inhalation profiles in dry powder inhalers. Eur Respir J 2001; 18:780-3. 51. Pedersen S. Inhaler use in children with asthma. Dan Med Bull 1987;34:234-49. 52. Brown PH, Ning AC, Greening AP, et al. Peak inspiratory flow through Turbuhaler in acute asthma. EurRespir J 1995;8:1940-1. 53. Clark AR, Hollingworth AM. The relationship between powder inhaler resistance and peak inspiratory conditions in healthy volunteers: implications for in vitro testing. J Aerosol Med 1993; 6: 99-110. 54. Srichana T, Martin GP, Marriott C. Dry powder inhalers: the influence of device resistance and powder formulation on drug and lactose deposition in vitro. Eur J Pharm Sci 1998; 7: 73-80. 55. Tarsin WY, Pearson SB, Assi KH, Chrystyn H. Emitted dose estimates from Seretide Diskusans Symbicort Turbuhaler following inhalation by severe asthmatics. Int J Phar 2006; 316: 131-7. 56. Pedersen S, Steffensen G. Fenoterol powder inhaler technique in children: influence of respiratory flow rate and breath-holding. Eur J Respir Dis 1986;68:207-14. 57. Pedersen S, Frost L, Arnfred T. Errors in inhalation techniques and efficiency in inhaler use in asthmatic children. Allergy 1986;41:118-24. 58. Borgstrom L, Asking L, Lipniunas P. An in vivo and in vitro comparison of two powder inhalers following storage at hot/humid conditions. J Aerosol Med 2005; 18: 304-10. 59. Robbins RA, Thomas AR, Proctor LM, Hoyt JC, Hayden JM. Heat decreases formoterol delivery. Chest 2005; 128: 4036-40. 60. Miraglia Del Giudice M, Decimo F, Capristo C. La terapia inalatoria in pediatria: pMDI. Pneumologia Pediatrica 2003; 12: 36-42. 61. Pauwels R, Newman S, Borgstrom L. Airway deposition and airway effects of antiasthma drugs delivered from metered-dose inhalers. Eur Respir J 1997; 109: 2127-38. 62. Ram FS, Wright J, Brocklebank D, White JE. Systematic review of clinical effectiveness of pressurised metered dose inhalers versus other hand held inhaler devices for delivering beta (2) agonists bronchodilators in asthma. BMJ 2001; 323: 901-5. 63. Brocklebank D, Wright J, Cates C. Systematic review of clinical effectiveness of pressurised metered dose inhalers versus other hand held inhaler devices for delivering corticosteroids in asthma. BMJ 2001; 323: 896-900. 64. Newman SP. Principles of metered-dose inhaler design. Respir Care 2005;50:1177-90. 65. Hess DR. Metered dose inhalers and dry powder inhalers in aerosol therapy. Respir Care 2005;50:1376-82. 66. O'Callaghan C, Barry P. Spacer devices in the treatment of asthma. BMJ 1997; 314: 1061-1062. 67. Prahl P, Jensen T. Decreased adreno-cortical suppression utilizing the Nebuhaler for inhalation of steroid aerosols. Clin Allergy 1987;17:393-8. 68.Salzman GA, Pyszczynsky DR. Oropharingeal candidiasis in patient treated with beclomethasone dipropionate delivered by metered-dose inhaler alone and with Aerochamber. J Allergy Clin Immunol 1988;81:424-8. 69. Newman SP, Clark AR, Talaee N, Clarke SW. Pressurized aerosol deposition in the human lung with and without an open spacer device. Thorax 1989; 44: 706-710. 70. O'Callaghan C. Delivery systems: the science. Pediatr Pulmonol 1997;15:51-4. 71. Novembre E, Monaco MG, Mori F, Calogero C. Gli spaziatori nel trattamento dell'asma bronchiale del bambino. Pneumologia Pediatrica 2003; 12: 43-8. 72. Leach CL, Davidson PJ, Boudreau RJ. Improved air way targeting with the CFC-free HFA-beclomethasone metered-dose inhaler compared with CFC-beclomethasone. Eur Respir J 1998; 12: 1346-53. 73. Leach CL, Davidson PJ, Hasselquist BE, Boudreau RJ. Lung deposition of hydrofuoroalkane-134a beclomethasone is greater than the of chlorofluorocarbon fluticasone and chlorofluorocarbon beclomethasone: a cross-over study in healthy volunteers. Chest 2002; 122: 510-6. 74. Menzies D, Nair A, Hopkinson P, McFarlane L, Lipworth BJ. Differential anti-inflammatory effects of large and small particle size inhaled corticosteroids in asthma. Allergy 2007; 62: 661-7. 75. Janssens HM, de Jongste JC, Hop WC, Tiddens HA. Extra- fine particles improve lung delivery of inhaled steroids in infants: a study in an upper airway model. Chest 2003; 123: 2083-8. 76. Sheth K, Wasserman RL, Lincourt WR, Locantore NW, Carranza-Rosenzweig J, Crim C. Fluticasone propionate/ salmeterol hydrofluoroalkane via metered-dose inhaler with integrated dose counter: performance and patient satisfaction. Int J Clin Pract 2006; 60: 1218-24. 77. Rubin BK, Durotoye L. How do patients determine that their metered-dose is empty? Chest 2004; 126: 1134-7. 78. Usmani OS, Biddiscombe MF, Barnes PJ. Regional lung deposition and bronchodilator response as a function of beta-2 agonist particle size. Am J Respir Crit Care Med 2005;172:1497-504. 79. Vanden Burgt JA, Busse WW, Martin RJ et al. Efficacy and safety overview of a new inhaled corticosteroid, QVAR (hydrofluoroalkane-beclomethasoneextrafine inhalation aerosol), in asthma. J Allergy Clin Immunol 2000;106:1209-26. 80. Geller DE. New liquid aerosol generation devices: systems that force pressurized liquids through nozzles. Respir Care 2002 Dec;47 (12) :1392-404; discussion 1404-5. 81. Hodder R, Price D. Patient preferences for inhaler devices in chronicobstructive pulmonary disease: experien
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
 Direttore scientifico Carmelo Salpietro
Direttore scientifico Carmelo Salpietro