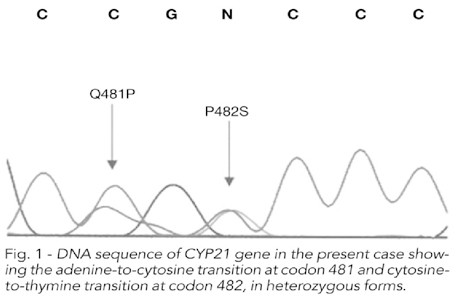
Rivista Italiana di Genetica e Immunologia Pediatrica Italian Journal of Genetic and Pediatric Immunology Quadrimestrale di aggiornamento scientifico dell'Euromediterranean Paediatric Foundation
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Decodificazione del linguaggio cerebrale Ipotesi per la trasmissione di informazioni tra cervello e calcolatore
Giuseppe Micali
Abstract Looking on to the dawn of the future, trying to know by intuition to foretell mathematically rigorous and logical concepts, is an ambitious and complex deed, but I think that it is anyway destined to lay the foundations for a future exploration and to produce a new scientific knowledge. The study on the historical and technological evolution of the language by conventions, the study on the history of the development of the printing techniques of the images, of the visual perception and the professional utilization of an Apple Macintosh Computer, have led me to a theory, essentially based on logic, which anyhow I want to explain in a logical hypothesis, although aware of its limitation. La notizia della presente teoria fu data dal Giornale di Sicilia il 13 novembre 1990. Questo lavoro, è stato pubblicato negli atti del World Congress on Biomedical Communications, Orlando-Florida 18 - 23 giugno 1994, dal quindicinale "Asis news", dalla rivista " Parentesi" di Messina, negli atti del Sesto Seminario di Medicina e Personal Computer (19-20 maggio 1993) -Università Cattolica del Sacro Cuore - Facoltà di Medicina e Chirurgia "Agostino Gemelli" - Roma Editrice: La Rivista Medica Italiana, "Progettare il futuro" 15-17 maggio 1997, PalAffari-Laboratorio di Ricerca Educativa Università di Firenze (Facoltà di Scienze Matematiche Fisiche e naturali). Il "World Congress on Biomedical Communications" è stato un eccezionale evento storico che non ha avuto precedenti nella storia della comunicazione multimediale biomedica.
Affacciarsi sulla soglia del futuro, cercare di intuire, per poter predire concetti matematicamente rigorosi e logici, è un'impresa ambiziosa e complessa, ma, credo, sia comunque destinata a gettare le basi per una futura esplorazione e per produrre nuove conoscenze scientifiche. Lo studio sull'evoluzione storica e tecnologica del linguaggio per convenzioni, lo studio sulla storia dello sviluppo delle tecniche di impressione delle immagini, della percezione visiva e l'utilizzazione professionale di un personal computer, mi ha condotto ad una teoria, fondata essenzialmente sulla logica, che desidero esporre consapevole della limitazione ad ipotesi ragionata. L'animale Uomo scoprà, col passare dei secoli, che il suo corpo era avvolto da tanti fili, ai quali diede il nome di nervi, collegati a una stazione di smistamento, che chiamà³ cervello. Piຠtardi conobbe l'elettricità e, con l'evoluzione tecnologica, l'applicà³ anche a scopo terapeutico, chiamando lo studio di questi fenomeni "elettrologia". La tecnica moderna che permette di misurare gli impulsi elettrici delle cellule nervose è basata sullo "Squid", uno strumento in grado di misurare campi magnetici estremamente piccoli, dalle dimensioni fino a un miliardesimo rispetto a quelle del campo magnetico della terra. I segnali vengono analizzati e trasformati in immagini da un calcolatore. In questo modo è possibile evidenziare la distribuzione e l'evoluzione nel tempo dell'attività elettrica in una particolare zona del cervello. Questa tecnica, da me descritta e pubblicata nell'anno 1990, è stata messa a punto l'anno successivo in Germania dall'ospedale di Amburgo in collaborazione con la Philips, e le prime immagini dell'attività elettrica del cervello sono diventate una "biblioteca di riferimento" destinata a diventare la base per la diagnosi di alcune forme di malattie nervose. Altre persone stanno sperimentando e studiando le meravigliose possibilità del computer sul corpo umano. La rivista Italiana "Virtual" nel 1990 informava che il dottor Jerrold S. Petrofsky a Irvine-California aveva inventato una nuova tecnica di riabilitazione basata sull'uso del computer, un minuscolo apparecchio allacciato in vita, facendo da collegamento tra cervello e arti, è in grado di ricevere, per mezzo di sofisticati sensori, gli impulsi cerebrali e ritrasmetterli agli arti, ordinando i movimenti. Il dottor Dave Warner a Loma Linda-Los Angeles sta cercando di trapiantare nel mondo della medicina l'uso di applicazioni avanzate di sintesi grafica e immersione in realtà virtuale. Biomuse è un sistema creato dalla Biocontrol System di Palo Alto e consiste in un processore in grado di elaborare impulsi elettrici di bassissima intensità , che capta l'attività elettromuscolare e la trasmette ad un computer. Questi impulsi bioelettrici, prodotti dai muscoli del paziente, sono captati a loro volta da elettrodi cutanei. Per mezzo di questo sistema si è in grado di percepire anche input dati dall'elettroencefalogramma. E', perciò, sempre più frequente l'approccio uomo-macchina, uomo-computer, uomo-elettricità per diagnosticare anomalie del nostro corpo, per effettuare accertamenti clinici e, a scopo terapeutico, con l'invio di segnali elettrici. Il computer è il nuovo domestico elettrico, il nuovo strumento di esplorazione scientifica al servizio della scienza, il quale consente di analizzare in modo del tutto nuovo le informazioni, offre nuovi parametri di valutazione e permette di giungere a risultati logici un tempo impossibili. Le mie teorie riguardano solamente l'ipotesi di una regia visiva, con manipolazione grafica di una mappa elettronica del corpo umano raffigurante i circuiti conduttori di energia, organizzata per la ricezione e trasmissione di impulsi elettrici. Questa configurazione elettronica alla quale ho dato il nome di "Electron", dovrebbe essere composta da una fusione di strumenti e tecniche già esistenti, essenziali per l'input dei dati corporei: -casco e DataGlove per l'immersione in realtà virtuale per ottenere il completo rilassamento del paziente; -sei telecamere ad ampio spettro, in similitudine al metodo aerofotogrammetrico, per rilevare le differenze strutturali esistenti nelle varie superfici del corpo; -esame ecografico totale per la misurazione di tutte le profondità corporee cosà come si fa in oceanografia con il sonar; -dati T.A.C.; -elettromiografia totale per la registrazione grafica dell'attività elettrica dei muscoli; -dati DNA per l'identificazione genetica del file sottoposto all'esame; -Squid; -Biomuse. Otterremmo, così, qualsiasi informazione sui circuiti conduttori di energia e una visualizzazione particolareggiata dell'intero apparato corporeo. Electron, perciò, dovrebbe essere composto da un hardware con interfacce sensoriali, integrato da un software con immersione in Realtà Virtuale con rendering di una mappa elettronica del corpo umano. In un caso di trombosi venosa ad esempio, facendo penetrare, con precisione senza precedenti, degli aghi simili a quelli per l'agopuntura -collegati opportunamente per l'output-, si potrebbe inviare energia elettrica perfettamente calibrata con il risultato di una frantumazione dei trombi senza che il paziente sia sottoposto al fastidio dell'anestesia e dell'intervento chirurgico. Durante un lungo percorso storico, l'Uomo lasciò la propria impronta comunicativa per fissare il suo pensiero e per stabilire l'iter delle convenzioni gestuali, foniche e grafiche, necessarie per registrare, conservare, tramandare e comunicare a distanza. Nel campo medico la ComputerGrafia è la fusione di molteplici convenzioni che permettono di esprimere chiaramente e più rapidamente la ricezione e la trasmissione dei dati. Il linguaggio scientifico, al giorno d'oggi, è costituito da una elaborazione di dati numerici codificati e descritti in forme geometriche. La ricezione di un elettroencefalogramma, ad esempio, è visualizzata da una grafia, corrispondente ad una modulazione elettrica, composta da segni convenzionali, nella quale ogni linea equivale ad un simbolo standard o ad una deformazione. In questo caso il segnale elettrico è un alfabeto, un linguaggio codificato per convenzioni. La principale attività del cervello è quella di inviare segnali elettrici a tutto il corpo umano. La decodificazione di queste scariche in dati-output condurrebbe probabilmente ad una decifrazione e ad un linguaggio comune. Codificare questi segnali in numeri significherebbe poter comunicare con il cervello e poter ricostruire elettricamente l'identico segnale? Una regola matematica elementare dice: "cambiando l'ordine dei fattori, il prodotto non cambia". Percià³ se è possibile ricevere impulsi elettrici dal cervello, sarà altrettanto possibile farli tornare indietro utilizzando un computer. Parecchi anni fa digitalizzaii una piccola parte di un tracciato elettroencefalografico e studiai il file in programmazione, progettando anche un software in grado di editare e analizzare la sua struttura informatica. Uno strano listato si presentà³ nel monitor. Intuii che cià³ che osservavo poteva essere la clonazione della grafia in forma di istruzioni alfanumeriche, necessarie al computer per una calibrata emissione. Per avere conferma sulla mia ipotesi, cancellai un numero da quel listato e mandai in stampa il file contenente la grafia del tracciato; il foglio che uscà dalla mia stampante laser era completamente bianco. Ritornai nel listato e cominciai a cancellare diverse linee alfanumeriche; con mio stupore, vidi scomparire anche in video il tracciato elettroencefalografico. Cancellando quei dati avevo annullato la struttura informatica e le istruzioni in linguaggio macchina, necessarie per la visualizzazione e per l'output. Il computer consente certamente una suprema definizione di dati ed è il mezzo primario per ottenere la registrazione del segnale neuroelettrico con perfetta densità e calibrazione. Percià³, se l'input del tracciato elettroencefalografico avvenisse in Electron, automaticamente, potremmo ottenere una clonazione pura del segnale elettrico-cerebrale. Si potrebbe, poi, iniziare un iter di convenzioni, fondato su un principio naturale di adattamento, cosà come fa il bambino quando si adatta agli schemi anche verbali già codificati dagli adulti. Se, per esempio, un tracciato è uguale all'intensità di dolore di uno spillo che punge un dito, si potrebbe ritrasmettere ad un altro cervello -usando dei nervi elettronici o gli stessi elettrodi di ricevimento (organizzati quale output del computer)- lo stesso dolore senza che avvenga alcuna puntura. Questo potrebbe essere l'inizio di una convenzione per stabilire un codice di linguaggio tra cervello e computer e per sintonizzarsi sulla "lunghezza d'onda" di una lingua neurologica, che permetterebbe un primitivo metodo di comunicazione tra macchina cerebrale e macchina elettronica. Considerazioni E' fuori dubbio che il computer invii segnali elettrici in output e che solamente un calcolatore elettronico è in grado di trasmettere tali segnali esattamente calibrati e alla perfetta densità . L'eventuale cavia, al contrario dell'elettroshock, sopporterebbe inequivocabilmente la scarica elettrica che sarebbe recepita esclusivamente come segnale e non come scossa. Il tracciato encefalografico direttamente in computer, garantirebbe l'input matematico delle coordinate del segnale elettrico. L'oggetto output, naturalmente, dovrebbe contenere le identiche caratteristiche dei 21 elettrodi convenzionali usati per l'input in modo inverso. Se l'input-output venisse dato sempre con la stessa configurazione elettronica, si potrebbe creare una "libreria di vocaboli", che, in realtà , corrisponderebbero ad un "codice alfabetico". Desidero mettere ancora in evidenza che, con il presente lavoro, ho inteso esporre solamente un ragionamento logico sul quale, in futuro, si potrebbero approfondire ulteriori studi: -sulla configurazione elettronica; -sull'interfaccia; -sugli elettrodi e sulle fibre elettroniche da usare; -sulla compatibilità ; -sui valori dei parametri di trasmissione; -sulle interfacce sensoriali per l'immersione nella Realtà Virtuale. Solo dopo aver sperimentato questi sistemi si potrà effettivamente stabilire la metodica-convenzione di comunicazione tra i due cervelli. Potremo: -accellerare la risposta comportamentale dell'Uomo? -ordinare alla cellula tumorale di non impazzire? -ordinare alla cellula di non invecchiare? Alle soglie del XXI secolo stiamo vivendo una nuova mutazione tecnologia che sta trasformando i processi primitivi per trasmettere l'informazione e questa evoluzione ci porterà sicuramente a nuove conquiste. L'Input-Graphia è, senza dubbio, l'eccelsa evoluzione dell'alba preistorica, ma, certamente, l'era informatica che stiamo vivendo è la primitiva scintilla di un futuro del linguaggio ai confini della realtà .
Vent'anni dopo La svolta del progresso scientifico è iniziata con l'avvento delle tecnologie informatiche e, alle soglie del terzo millennio, molte sono state nel mondo le intuizioni tecnologiche che hanno prodotto innovazioni e strategie alternative in medicina. A due delle mie tre interrogazioni conclusive è stata data dalle nuove tecnologie scientifiche risposta affermativa. L'identità e la trasformazione della ricerca si costruisce anche nel raccontare le storie che viviamo o che abbiamo vissuto. Percià², sento il dovere di documentare con alcuni links parte degli sviluppi di questa avventura nell'impossibile iniziata vent'anni fa. • Tetraplegico muove gli oggetti con la forza del pensiero • Sedici anni fa anticipಠil futuro • Invia impulsi elettrici dagli occhiali al cervello • Mano cibernetica Anno 2008: il computer parla con l'uomo La sperimentazione del primo processo di riconoscimento verbale con il software "Adamo 9001", avvenuto con la mia voce il 24 luglio 2008 a San Giorgio di Gioiosa Marea (Messina), ha sviluppato l'atto del "contatto", il primo impulso elettrico-vocale tra uomo e macchina. Alle spalle, un elaborato linguaggio di programmazione del processo di inserimento della parola sonora e della ricezione-risposta del programma, assieme alla capacità di apprendimento di eventuali audiorisposte tramite qualsiasi voce. Un piccolo tassello aggiunto al puzzle dell'intelligenza artificiale senza la presunzione di aver potuto dare al software la capacità di pensare, una ulteriore dimostrazione che la fantascienza puಠdivenire realtà . Sono fermamente convinto che dopo che avremo imparato la grammatica e la sintassi del "sistema uomo", potremo comunicare col cervello utilizzando lo stesso sistema di telegrafia senza fili inventato da Guglielmo Marconi. La decodificazione del meraviglioso "libro della vità " è appena iniziata.
"I computer sono incredibilmente veloci, accurati e stupidi. Gli uomini sono incredibilmente lenti, inaccurati e intelligenti. Insieme sono una potenza che supera l'immaginazione." Albert Einstein
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
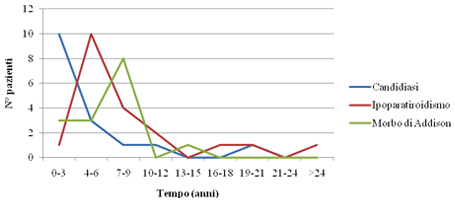
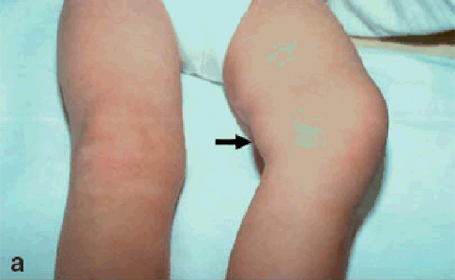
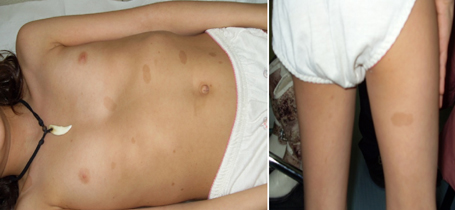
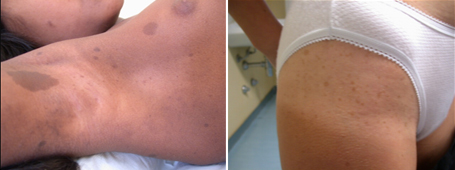
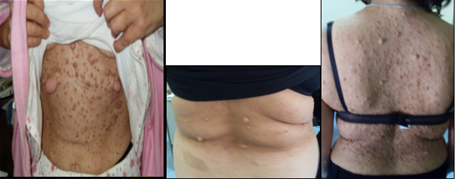
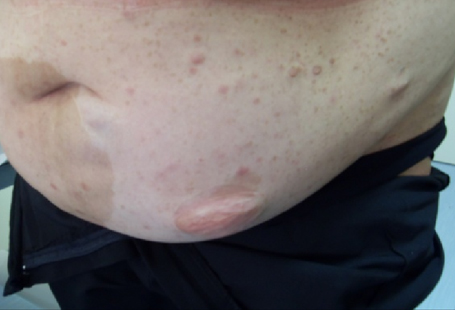
Figura 1: età d'esordio della triade sintomatologica classica dell'APECED La candidiasi mucocutanea insorge con un picco massimo nel primo anno di vita. Successivamente, in ordine cronologico, compare l'ipoparatiroidismo, con un picco verso i 6 anni di età . Infine il morbo di Addison si manifesta con la massima incidenza verso i 9 anni. Bibliografia 1. Heino M, Peterson P, Kudoh J, Shimizu N, Antonarakis S, Scott H and Krohn K. APECED mutations in the autoimmune regulator (AIRE) gene. Hum Mutat 2001;18:205-211. 2. Nagamine K, Peterson P, Scott S. H, Kudoh J, Minoshima S, Heino M, Krohn K. J. E, Lalioti M. D, Mullis P. E, Antonarkis S. E, Kawasaki K, Asakawa S, Ito F, Shimizu N. Positional cloning of the APECED gene. Nature Genetics. 1997. 17: 393-403. 3. Su MA et Anderson MS. AIRE: an update. Curr Opin Immunol 2004;16:746-752. 4. Peterson P, Org T, Rebane A. Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol. 2008;12: 948-957. 5. Badolato R et Rosatelli MC. La poliendocrinopatia autoimmune di tipo 1. Prospettive in pediatria 2002;32:313-318 6. Pradeep G.K, Malini Laloraya, Jin-Xiang She. Population Genetics and functions of the autoimmune regulator (AIRE). Endocrinol Metab Clin N Am. 2002. 31: 321-338. 7. Kuroda N, Mitani T, Takeda N Ishimaru N, Arakiki R, Hayashi Y, Bando Y, Izumi K, Takahashi T"e;¦and Matsumoto M. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of AIRE-deficient mice. J Immunol 2005;174:1862-1870. 8. Alimohammadi M, Bjà¶rklund P, Hallgren A, Pà¶ntynen N, Szinnai G, Shikama N, Keller M, Ekwall O, Kinkel S, Husebe E, Gustafsson J, Rorsman F, Peltonen L, Betterle C, Perheentupa J, Ǻkerstrà¶m G, Westin G, Scott H, Hollà¤nder G, Kà¤mpe O. Autoimmune Polyendocrine Syndrome Type 1 and NALP5, a Parathyroid Autoantigen. N Engl J Med 2008;358:1018-28. 9. Sato K, Sato U, Tateishi S, Kubo K, Horikawa R, Mimura T, Yamamoto K, Kanda H. AIRE down regulates multiple molecules that have contradicting immune-enhancing and immune-suppressive functions. Biochemical and Biophysical Research Communications. 2004. 318: 935-940. 10. Cavadini P, Vermi W, Facchetti F, Fontana S, Nagafuchi S, Mazzolari E, Sediva A, Marrella V, Villa A, Fischer A, Notarangelo Ld, Badolato R. AIRE deficiency in thymus of 2 patients with Omenn syndrome. JCI 2005;115 (3) :728-732. 11. Vogel A, Strassburg C, Obermayer-Straub P, Brabant G and Manns MP. The genetic background of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy and its autoimmune disease components. J Mol Med 2002;80:201-211. 12. Dittmar M et Kahaly Gj. Polyglandular Autoimmune Syndromes: Immunogenetics and Long-Term Follow-Up. JCEM 2003;88 (7) :2983-2992. 13. Tazi-Ahnin R, Mcdonagh AJG, Wengraf DA, Lovewell TRJ, Vasilopoulos Y, Messenger AG, Cork MJ, Gawkrodger DJ. The autoimmune regulator gene (AIRE) is strongly associated with vitiligo. BJD 2008;1-6. 14. Betterle C, Zanchetta R. Update on autoimmune polyendocrine sindrome (APS). Acta Bio Medica 2003;74:9-33. 15. Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature 2005;435:584-588. 16. Gylling M, Kà¤à¤rià¤inen E, Và¤isà¤nen R, Kerosuo L, Solin Ml, Halme L, Saari S, Halonen M, Kà¤mpe O, Perheentupa J, Miettinen A. The Hypoparathyroidism of Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy Protective Effect of Male Sex. JCEM 2003;88 (10) :4602-4608. 17. Gavanescu I, Benoist C, Mathis D. B cells are required for AIRE-deficient mice to develop multi-organ autoinflammation: a therapeutic approach for APECED patients. PNAS 2008; 105 (35) :13009-13014. Indirizzo per la corrispondenza Professor Raffaele Badolato Clinica Pediatrica dell'Università di Brescia Piazzale Spedali Civili 1 - 25123 Brescia Tel. 0303995717 - e-mail: badolato@med.unibs.it
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
L'esofagite eosinofila in età pediatrica Eosinophilic esophagitis in children
Valeria Ferraà¹, Paolo Rossi, Annalisa Famiani, Antonella Talenti, Italia Loddo, Claudio Romano Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina
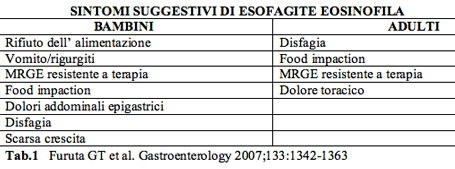
Abstract During the last decade, clinical practice saw a rapid increase of patients with esophageal eosinophilia who were thought to have gastroesophageal reflux disease but who did not respond to medical and/or surgical management. Subsequent studies demonstrated that these patients had a new disease termed eosinophilic esophagitis (EE). EE is a clinicopathological disease characterized by clinical symptoms of esophageal dysfunction, "e;¥ 15 eosinophils/HPF, exclusion of other disorders with similar clinical, histological or endoscopic features, especially GERD (use of high dose PPI treatment or normal Ph monitoring). Appropriate treatments include dietary approaches based eliminating exposure to food allergens, topical or systemic corticosteroids, cromolyn sodium and biologics. Riassunto Negli ultimi dieci anni si ਠassistito ad un progressivo incremento dei pazienti sia di età adulta che di età pediatrica affetti da una nuova patologia: l'esofagite eosinofila (EE). L'EE ਠun disordine clinico-patogenetico ad eziologia ancora sconosciuta, presumibilmente immuno-allergica, caratterizzato da: sintomi gastrointestinali, biopsia esofagea che mostri "e;¥ 15 eosinofili/HPF, assenza di MRGE, intesa come ph-metria negativa e/o mancata risposta ad alte dosi di inibitori di pompa protonica (2 mg/Kg/die). La terapia dell'EE piuttosto complessa, ਠvolta al mantenimento della remissione clinica e alla prevenzione delle complicanze, oltre che all' approccio della fase acuta. Le possibili opzioni terapeutiche attualmente includono: diete di eliminazione, steroidi per via sistemica o topica, sodio cromoglicato. I composti biologici sono ancora in fase di studio. Introduzione L'esofagite eosinofila (EE) ਠun entità clinica caratterizzata da una severa eosinofilia esofagea con iperplasia squamosa epiteliale, che generalmente si manifesta con sintomi gastrointestinali alti, in primis esofagei. E' una patologia emergente la cui prevalenza ha subito un netto aumento negli ultimi dieci anni. Ogni età puಠessere interessata, l'esordio pi๠comune ਠnell'infanzia e nell'adolescenza. Pi๠precisamente ਠstato dimostrato vi siano due picchi di incidenza: nel bambino tra i 6 e gli 8 anni, nell'adulto intorno alla terza-quarta decade di vita. L'incidenza dell'EE ਠdi 1/10.000 casi l' anno, con una prevalenza di 4/10.000. E' pi๠frequente nel sesso maschile (rapporto M/F di circa 3:1). E' una condizione clinica ad eziopatogenesi presumibilmente immuno-allergica. Sono stati chiamati in causa diversi meccanismi che suggeriscono una disregolazione immunologica e il contributo di allergeni sia alimentari che inalanti. Fenomeni autoimmunitari che coinvolgono altri tratti dell'apparato gastrointestinale possono essere implicati nello sviluppo di questa malattia. In oltre la metà dei pazienti viene riferita una storia familiare o personale positiva per allergia con episodi di asma, rinite o dermatite atopica. Il meccanismo di reclutamento degli eosinofili ਠregolato da numerosi mediatori dell' infiammazione che includono citochine pro-infiammatorie (IL3-IL4-IL5), chemochine (RANTES) e proteine pro-macrofagiche come le eotaxine 1, 2, 3. Solo l'IL5 e le eotaxine sono perಠdotate di elevata specificità nei confronti degli Eosinofili, in particolar modo l' eotaxina 1 possiede un ruolo chiave nella modulazione dell' infiltrato eosinofilo a livello del tratto gastrointestinale. Altro ruolo fondamentale nella genesi dell'EE ਠsvolto dalla citochina Th2 e dall'IL-5 necessari nell'induzione di tale patologia. E' stato dimostrato infatti che una iperespressività dell'IL-5 sotto il controllo delle cellule T (CD2) o degli enterociti intestinali puಠdeterminare un aumento della concentrazione di eosinofili a livello della mucosa esofagea. Quadri clinici Clinicamente l'esofagite eosinofila puಠpresentarsi con una varietà di quadri sintomatologici. Nei bambini di età inferiore ai 5-6 anni il quadro clinico ਠin genere caratterizzato da: dolore addominale in sede epigastrica, inappetenza, episodi di vomito e/o rigurgiti. Nel bambino pi๠grande ed in età adolescenziale predominano invece la disfagia, il dolore toracico e il "e;food impaction"e;.
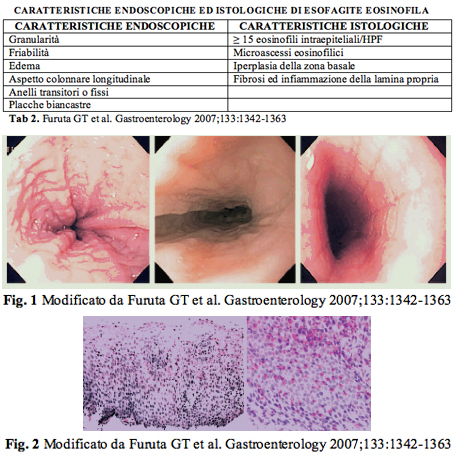
Diagnosi L'esofagogastroduodenoscopia (EGDS) con biopsie esofagee multiple puಠessere considerata la tecnica diagnostica gold standard. Le biopsie vanno eseguite a livello di tutto l'esofago (prossimale, medio, distale) e a livello gastroduodenale per escludere un concomitante quadro di gastroenteropatia eosinofila. E' utile eseguire almeno 4-5 prelievi bioptici (1-2 per porzione esofagea) per raggiungere una sensibilità diagnostica pari al 90-95%. L'aspetto macroscopico dell'esofago dipende dal grado e dall' entità dell'infiltrato eosinofilo presente. Il quadro endoscopico puಠessere caratterizzato da granularità , friabilità , edema, aspetto colonnare longitudinale, anelli transitori o fissi. Tale condizione si accompagna ad importanti alterazioni della motilità e peristalsi esofagea e giustifica la disfagia che talvolta rappresenta il "e;sintomo guida"e;. Il pattern di infiltrazione puಠessere presente nella porzione sia prossimale che distale dell' esofago. La presenza di placche biancastre che sormontano la mucosa esofagea possono essere evidenti nelle fasi pi๠avanzate, necessitano di diagnostica differenziale nei confronti di lesioni da Candida e sono espressione della presenza di ascessi eosinofili. Sebbene nessuna di queste caratteristiche possa essere considerata patognomonica di EE, uno o pi๠dei suddetti aspetti endoscopici possono essere fortemente suggestivi di tale patologia, in presenza di sintomi clinici. La stenosi o la riduzione del calibro esofageo rappresentano le lesioni pi๠gravi ed avanzate, secondarie all'infiammazione a prevalente componente eosinofila, alla deposizione di collagene e alla conseguente fibrosi. Tale condizione si associa al rischio di episodi di bolo carneo che talvolta rappresentano una reale complicanza o emergenza. Non esistono dati certi in letteratura che permettano di individuare un'istologia tipica dell' esofagite eosinofila, ovvero il cut-off nel conteggio degli eosinofili nella biopsia esofagea. I dati disponibili indicano che un numero di eosinofili "e;¥ 15 per campo (HPF) rappresenti la diagnosi istologica di EE da correlare poi al quadro clinico. Altre caratteristiche istologiche utili, ma non indispensabili per la diagnosi istologica sono: l'iperplasia della zona basale, l'allungamento delle papille, la fibrosi della lamina propria e la stratificazione in superficie degli eosinofili con aggregati o microascessi. Effettuata la diagnosi clinica ed istologica di EE ed inquadrato il paziente dal punto di vista anamnestico (familiarità o anamnesi personale immunoallergologica) ਠpossibile effettuare un ciclo di terapia con IPP per 8 settimane, verificandone la risposta, oppure una ph-impedenzometria delle 24 ore. Contemporaneamente il paziente deve essere introdotto nell'iter diagnostico allergologico, per l'identificazione di un possibile allergene implicato (sia trofo che aereo allergene). Molti studi infatti, sembrano confermare come l'allergia alimentare giochi un ruolo significativo nella sua patogenesi. L'ipotesi di una reazione di ipersensibilità di tipo I verso alcuni alimenti ਠsupportata dal riscontro di eosinofilia sia a livello tissutale che a livello sierico, dagli elevati livelli di IgE e da una risposta positiva alla terapia steroidea. Nei soggetti sensibilizzati si ਠriscontrata la presenza di una connessione tra le IgE e i siti recettoriali dei mastociti per il frammento Fc; si potrebbe quindi supporre che il contatto di queste cellule con specifici antigeni alimentari possa provocarne la degranulazione, con il conseguente rilascio di alcuni fattori citoplasmatici quali il fattore chemiotattico eosinofilo ed il fattore di attivazione piastrinico, in grado di attirare a loro volta gli eosinofili in queste sedi. Negli adulti con EE solo occasionalmente l'allergia alimentare rappresenta un inequivocabile fattore patogenetico, viceversa puಠrivestire un ruolo fondamentale in età pediatrica. Si procederà pertanto alla determinazione delle IgE sieriche e degli eosinofili circolanti, all'esecuzione degli skin prick test e skin patch test verso trofo e aereo allergeni, e al dosaggio delle IgE specifiche (RAST) verso gli allergeni risultati positivi ai prick.
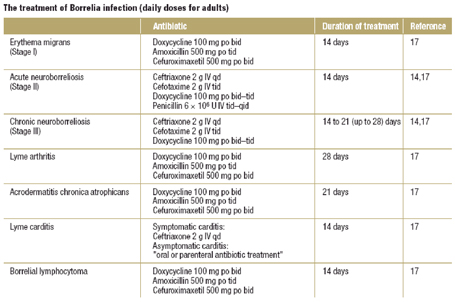
Terapia Il trattamento dell'EE resta tuttora controverso e assai complesso. Il ruolo primario della terapia rimane l'approccio della fasi acute e la prevenzione delle complicanze, oltre che il mantenimento della remissione clinica. I cardini dell' approccio terapeutico nella EE sono costituiti da dieta di eliminazione, corticosteroidi per via sistemica o topica, IPP (Inibitori di pompa protonica), sodio cromoglicato, antileucotrienici (Montelukast) e anticorpi monoclononali anti-IL5 (Mepolizumab). La terapia farmacologica va valutata in base all'andamento clinico del paziente, alle alterazioni anatomiche e al grado di interessamento flogistico istologico. L'identificazione degli antigeni alimentari causali tramite prick test e patch test e la loro successiva eliminazione puಠdeterminare un miglioramento sia clinico che istologico. Nei pazienti con meno di 10 allergeni identificati si puಠavviare una dieta di eliminazione oligoantigenica per 3 mesi, nei pazienti invece, con positività superiore ai 10 allergeni puಠessere proposta una dieta elementare per 8 settimane con successiva reintroduzione graduale degli alimenti, seguita da controlli endoscopici seriati per mettere in evidenza eventuali alterazioni esofagee macroscopiche e/o istologiche. L'eliminazione degli allergeni alimentari sospetti in associazione all'uso di IPP puಠessere considerato il trattamento di elezione. In alternativa ਠpossibile ricorrere all'uso di steroidi per via sistemica o topica, molto efficaci nell' indurre una remissione clinica e nel ridurre l'eosinofilia mucosale esofagea. Tuttavia questa remissione risulta essere temporanea con precoce ricaduta alla sospensione della terapia (circa 4-6 mesi). Gli steroidi sistemici (prednisone o metilprednisolone) sono generalmente utilizzati nelle fasi acute di malattia per un ciclo di 2-4 settimane, mentre gli steroidi topici (fluticasone dipropionato spray, budesonide sospensione orale) nel controllo a lungo termine della sintomatologia. Gli IPP vengono in genere utilizzati in fase diagnostica o in corso di terapia con corticosteroidi per via sistemica. Sono stati riportati inoltre casi di pazienti in età adulta, che rispondono alla terapia con sodio cromoglicato. Questo farmaco in genere ben tollerato nella terapia a breve termine, previene il rilascio di mediatori tossici da parte dei mastociti, come l'istamina, il fattore attivante le piastrine e i leucotrieni. Infine prospettive terapeutiche che attendono di essere valutate adeguatamente mediante studi clinici randomizzati sono costituiti dall'utilizzo di anticorpi monoclonali anti-IL5 o degli antileucotrienici. Per quanto riguarda l'anti-IL5 (Mepolizumab) ਠstato dimostrato in diversi studi un'azione sinergica con i glucocorticoidi in grado di ridurre significativamente il numero degli eosinofili nel sangue periferico. Il montelukast, invece, si ਠdimostrato molto efficace nel controllo dei sintomi clinici e potrebbe costituire un' alternativa al trattamento a lungo termine con corticosteroidi. Questo farmaco sembrerebbe in grado di inibire selettivamente il recettore del cisteinil leucotriene D4 presente sulla superficie degli eosinofili, con conseguente inibizione del reclutamento degli eosinofili stessi. Bibliografia 1. Furuta GT, Liacouras CA, Collins MH et al. Eosinophilic esophagitis in children and adults : a systematic review and consensus raccomendations for diagnosis and treatment. Gastroenterology 2007;133:1342-1363 2. De Angelis P, Markowitz JE, Torroni F et al. Pediatric eosinophilic esophagitis:towards early diagnosis and best treatment. Digestive and liver disease 2006;38:245-51 3. Gupte AR, Draganov PV et al. Eosinophilic esophagitis. World J Gastroenterol 2009;7:17-24 4. Rothenberg ME, Mishra A, Collins MH et al. Pathogenesis and clinical features of eosinophilic esophagitis. J Allergy Clin Immunol 2001;108:891-894 5. Noel RJ, Putnam PE, Rothenberg ME et al. Eosinophilic esophagitis N Engl J Med 2004;351:940-1 6. Rothenberg ME et al. Eosinophilic gastrointestinal disorders J Allergy Clin Immunol 2004 ;113 :11-28 7. Liacouras CA Eosinophilic esophagitis in children and adults. J Pediatr Gastroenterol Nutr 2006;37 Suppl:S23-8 8. Cheung KM, Oliver MR, Cameron DJ et al. Esophageal eosinophilia in children with dysphagia Dig Dis Sci 2003;48:22-29 9. De Angelis P, Morino G, Pane A et al. Eosinophilic esophagitis: management and pharmacotherapy. Expert opin Pharmacother 2008;95:731-740 Review 10. Ferguson DD, Foxx-Orenstein E et al. Eosinophilic esophagitis: an update. Diseases of the esophagus 2007;20:2-8 11. Furuta GT, Straumann G et al. Review article:the pathogenesisi and management of eosinophilic esophagitis. Aliment Pharmacol Ther 2006;24:173-82 12. Gonsalves N, Policarpio M, Zhang Q et al. Histopathologic variability and correlates in adults with eosinophilic esophagitis Gastrointestin Endoscop 2006;64:313-319 13. Dahms BB et al. Reflux esophagitis:sequelae and differential diagnosis in infants and children including eosinophilic esophagitis Pediatr Dev Pathol 2004;7:5-16 14. Anmed A et al. A novel endoscopic appearance of idiopathic eosinophilic esophagitis. Endoscopy 2008, 32:S33 15. Lee RG. Marked eosinophilia in esophageal mucosal biopsies. Am J Surg Pathol 1995;9:475-479 16. Spergel JM, Andrews T, Brown-Whitehorn TF et al. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthsma Immunol 2005;95:336-343 17. Assa'ad AH, Putnam PE, Collins MH et al. Pediatric patients with eosinophilic esophagitis in children:successful treatment with oral corticosteroids J Pediatr Gastroenterol Nutr 2008;26:380-385 18. Noel RJ, Putnam PE, Collins MH et al. Clinical and immunopathologic effects of swallowed fluticasone for eosinophilic esophagitis. Clin Gastroenterol Hepatol 2004;2:568-575 19. Markowitz JE, Spergel JM, Ruchelli E et al. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol 2007;98:777-82 20. Teitelbaum et a. Eosinophilic esophagitis in children: immunophatological analysis and response to fluticasone propionate Gastroenterol 2002;122:1216-1225 21. Aceves SS, Dohil R, Newbury RO et al. Topical viscous budesonide suspension for treatment of eosinophilic esophagitis J Allergy Clin Immunol 2005;116:705-706 22. Attwood SEA, Lewis CJ, Bronder CS et al. Montelukast eosinophilic esophagitis: a novel treatment using. Gut 2003;52:181-185 23. Garrett JK, Jameson SC, Thomson B et al. Anti-Interleukin-5 therapy for hypereosinophilic syndromes. J Allergy Clin Immunol 2004;113:115-9 24. Schaefer ET, Fitzgerald JF, Molleston JP et al. Comparison of oral prednisone and topical fluticasone in the treatment of eosinophilic esophagitis:a randomized trial in children. Clin Gastroenterol Hepatol 2008;6:165-173
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Ereditarietà non HLA della celiachia Non-HLA inharitance of celiac disease
Valeria Ferraà¹, Annalisa Famiani, Paolo Rossi, Donatella Comito, Elisabetta Mazzola, Romina Gallizzi, Claudio Romano Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina
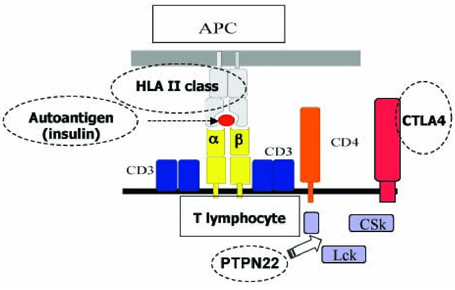
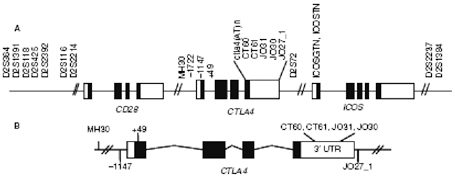
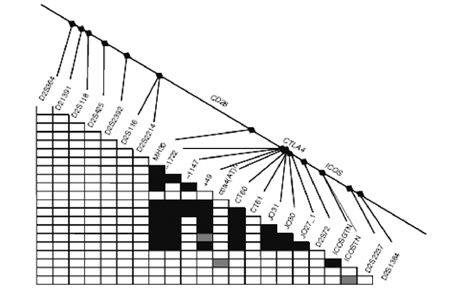
Fig. 1 Correlazione CTLA4/HLA II nella risposta autoimmune Numerosi altri geni candidati, fuori dal complesso HLA, sono stati testati ma ad oggi nessuno ਠrisultato associato in maniera certa e definitiva con la celiachia. Le strategie future per identificare eventuali geni predisponenti diversi dall'HLA potrebbero essere di 3 tipi: 1. selezionare i migliori geni candidati e testarli ognuno individualmente (approccio del gene candidato) 2. intraprendere uno studio sistematico di associazione sulle regioni identificate per linkage (genome-wide scan), come 5q31-33, utilizzando polimorfismi del singolo nucleotide (SNPs) 3. condurre studi di genome-wide association. Ad esempio con la strategia 1 sono già stati selezionati, testati ed esclusi i seguenti geni: transglutaminasi tissutale TG2, metalloproteinasi della matrice 1 e 3 (MMP1 e 3) che sembrano coinvolti nell'atrofia dei villi, geni del T cell receptor (a, b, g, d), IL12B che modifica l'espressione della citochina proinfiammatoria IL12, CD28, CD80 (B7-1) , CD86 (B7-2), KIR e LILR, cluster genici coinvolti nella funzione delle cellule T e NK. La strategia 2 del genome-wide linkage scan, nonostante tutti i noti limiti del metodo, ha avuto un buon successo, indicando un linkage ragionevolmente attendibile con la regione 5q31-33 (Greco et al, 1998). Tale dato ਠstato successivamente confermato in diversi studi indipendenti condotti su popolazioni del Nord e del Sud Europa. Attualmente, numerosi gruppi stanno procedendo allo studio sistematico dei geni candidati presenti in questa regione, che ਠcomunque molto estesa. La strategia 3 di genome-wide association study ਠquella che si ritiene pi๠promettente per rivelare i geni malattia associati debolmente, che sfuggono alla strategia precedente. Gli studi di linkage, mediante i quali si cerca di localizzare i geni-malattia sui cromosomi, forniscono per la celiachia risultati promettenti. Oltre a confermare la stretta associazione della malattia con la regione 6p21 (dove ਠlocalizzato il complesso HLA), forniscono evidenza di linkage con altri loci non-HLA, in particolare con le regioni 5q31-33 (CELIAC 2), 2q33 (CELIAC 3), 19p13 (CELIAC 4). Alcune di esse (CELIAC 2 e CELIAC 4) sono state anche descritte nelle malattie infiammatorie croniche intestinali (IBD5 e IBD6 rispettivamente), suggerendo almeno in parte una base comune di suscettibilità alla malattia. In particolar modo, mutazioni del gene IXB per la miosina (MYO9B), localizzato sul cromosoma 19p13, sembrerebbero correlate con un maggiore rischio di malattia celiaca, come evidenziato in uno studio condotto da un gruppo di ricercatori italiani.
Sono stati condotti anche numerosi studi su altri geni non-HLA classici presenti in questa regione cromosomica, in particolare quelli coinvolti nei processi immunitari e quindi, ragionevolmente, nella abnorme risposta al glutine. Purtroppo, un controllo inadeguato del linkage disequilibrium ha fatto emergere numerose associazioni, che successivamente non sono state confermate. I geni MICA, LMP2, TAP1, TAP2, DMA e DMB sono tutti esempi di loci riportati associati alla celiachia, ma nessuno di essi si ਠconfermato in studi in cui il linkage disequilibrium era adeguatamente controllato. Il gene MICA, in particolare, codifica per una proteina stress-indotta espressa sull'epitelio intestinale. La sua funzione ਠquella di legarsi ai linfociti T ï§ï¤ ampiamente espressi nelle lesioni intestinali tipiche della malattia celiaca. Il gene MICA possiede un trinucleotide (GCT) che si ripete nei segmenti transmembrana in aggiunta all'allele A5.1, generando una mutazione puntiforme che induce uno "e;stop"e; prematuro presumibilmente responsabile dei meccanismi di "e;disregolazione immunologica"e; in soggetti con celiachia. Altri studi di polimorfismo genico relativo ai geni di sintesi delle citochine infiammatorie, in particolare IL 4, IL13, IL 17B e IL 5, hanno dimostrato una possibile correlazione con la celiachia. Un altro approccio per cercare fattori genetici coinvolti nelle patologie multifattoriali ਠcostituito dall'analisi di associazione con geni candidati. Un'associazione significativa ਠstata riportata per il gene che codifica per la regione CELIAC 3 (gene CD28/CTLA4/ICOS) sul cromosoma 2q33. Diversi polimorfismi genici compresi in questa regione sembrano essere correlati anche con altre patologie autoimmuni, ad esempio il diabete tipo I, l'ipotiroidismo autoimmune o la sclerosi multipla.
Fig. 4 Polimorfismi genici del CD28/CTLA4/ICOS (CELIAC3) Per quanto concerne il TNF, il cui gene si trova nella Classe III dell'HLA, ci sono dati interessanti. Le citochine TNFα e β hanno un ruolo fondamentale nell'immunità mediata dalle cellule T ed ਠstata dimostrata una loro elevata espressione negli aplotipi B8-DR3. Nel 1996 McManus et al. riportavano associato alla celiachia un polimorfismo del gene TNF2 (TNF-308A), che costituisce un polimorfismo funzionale nel promotore di TNFα, aumentandone l'espressione. Tale dato ਠstato successivamente confermato in pazienti spagnoli (de la Concha et al, 2000). Un altro gene candidato studiato ਠstato IL12B, codificante la subunità p40 della citochina pro-infiammatoria IL-12. IL12B ਠstato selezionato come candidato in base alla sua funzione, perchਠmappa nella regione 5q31.1-33.1 e perchਠਠstata descritta un'associazione con il diabete di tipo 1 di un polimorfismo localizzato nella regione 3'UTR di questo gene. Lo stesso polimorfismo non ha mostrato associazione con la celiachia nelle popolazioni italiana e scandinava. Un'associazione con celiachia ਠstata anche esclusa per i geni che codificano la transglutaminasi tissutale, alcune peptidasi e i recettori dei linfociti T. In conclusione, la sempre pi๠numerosa variabilità genica identificata attraverso gli studi di genoma wide association e di linkage, in associazione con la sempre pi๠ampia conoscenza dei meccanismi patogenetici coinvolti nella celiachia permetterà , in un prossimo futuro, di ampliare la ricerca con l' intento di identificare loci genici non-HLA pi๠specifici, correlati con la suscettibilità alla malattia. Bibliografia 1. Petronzelli F, Bonamico M, Ferrante P, et al. Genetic contribution of the HLA region to the familial clustering of coeliac disease. Ann Hum Genet 2007; 61: 307-17 2. Sollid LM. Coeliac Disease: dissecting a complex inflammatory disorder. Nat Rev Immunol 2002; 2: 647-55 3. Karell K, Louka AS, Moodie SJ, et al. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the european genetics cluster on celiac disease. Hum Immunol 2003; 64:469-77 4. S.S. Amundsen, AT Naluai, H Ascher et al. Genetic analysis of the CD28/CTLA4/ICOS (CELIAC 3) region in coeliac disease. Tissue Antigens 2004;64:593-599 5. A.Latiano, B. Mora, M. Bonamico et al. Analysis of candidate genes on chromosomes 5q and 19p in celiac disease. JPGN 2007;45:180-186 6. A.M.Pagola, G. Perez de Nanclares, J.C Vitoria et al. No associaton of CTLA4 Gene with celiac disease in the basque population. JPGN 2003;37:142-145 7. A. H. Gudjonsdottir, S. Nilson, A. Torinsson et al. Association between genotypes and phenotypes in coeliac disease . JPGN 2009;49:165-169 8. P.M. Holopainen and J.A. Partanen Technical note: linkage disequilibrium and disease-associated CTLA4 Gene Polymorphisms. J. Immunol 2001;167:2457-2458 9. MI Torres, MA Lopez Casado, A Rios New aspect in celiac disease. World Journal Gastroenterol 2007; 28;13: 1156-1161 10. Ueda H, Hwson J, Esposito L et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease . Nature 2003;423.506-11 11. K. Haimla, T Smedberg, K. Mustalahti et al. Genetic association of celiac disease susceptibility to polymorphism in the ICOS gene on chromosome 2q33. Genes Immun 2004;5:85-92 12. Mora B, Bonamico M, Indovina P et al. CTLA4 49 A/G dismorphism in Italian patients with celiac disease . Hum Immunol 2003;64:297-301 13. Holopainen P, Mustalhati K, Uimari P et al. Candidate gene regions and genetic heterogeneity in gluten sensitivity . Gut 2001;48:696-701 14. Louka AS, Torinsson Naluai A, D'alfonso S et al. The IL-12B gene does not confer susceptibility to celiac disease Tissue Antigens 2002;59:70-2 15. Greco L, Percopo S, Clot F et al. Lack of correlation between genotype and phenotype in celiac disease. JPGN 1998;26:286-90 16. Amundsen SS, Monsuur AJ, Wapenaar MC et al. Association analysis of MYO9B gene polymorphisms with celiac disease in a Swedish/Norvegian cohort. Hum Immunol 2006;67:341-5 17. Grohn V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MCH molecules by intestinal ephitelial gammadelta T cells. Science 1998;279:1737-40 18. Rueda B, Pascual M, Lopez-Nevot MA et al. Association of MICA-A5.1 allele with susceptibility to celiac disease in a family study. Am J Gastroenterol 2003;98:359-62 19. Clot F, Fulchignoni-Lataud MC, Renoux C et al.Linkage and association study of the CTLA-4 region in celiac disease for Italian and Tunisian populations. TissueAntigens 1999:54:527-30 20. Shi Y, Ullrich SJ, Zhang J et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J Biol Chem 2008:275:19167-76
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Ghrelina: la molecola della fame Ghrelin: the molecule of hunger
Piera Vicchio, Annamaria Salpietro, Rosangela Caruso, Valeria Chirico, Caterina Grosso, Giovanna Elisa Calabrà², Maria Concetta Cutrupi, Silvana Briuglia, Caterina MunafಠDipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina
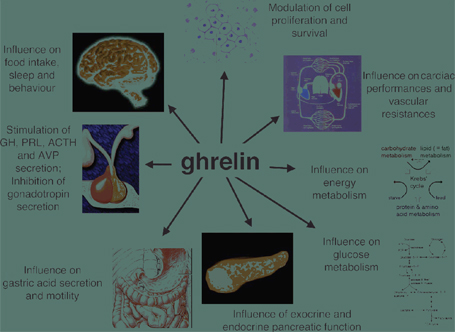
Riassunto La ghrelina ਠun ormone peptidico prodotto dallo stomaco, che presenta numerose attività neuroendocrine, metaboliche ma anche attività non endocrine. La scoperta relativamente recente di questa molecola ਠbilanciata da un grande interesse che gli studiosi hanno dimostrato per le sue molteplici attività biologiche. In questa breve review tratteremo gli aspetti biologici pi๠studiati fino a questo momento. Abstract Ghrelin is a peptide hormone produced by the stomach, which has numerous neuroendocrine, metabolic and non-endocrine activities. The relatively recent discovery of this molecule is balanced by a great interest that researchers have shown for its multiple biological activities. In this brief review will discuss the biological aspects studied to date. Introduzione La ghrelina ਠun ormone peptidico, scoperto nel 1999, costituito da 28 aminoacidi secreto soprattutto dalle cellule X/A della mucosa oxintica dello stomaco e che possiede numerose attività a livello del SNC ma non solo (1). Il termine ghrelina (GH + "e;release"e;) ricalca la sua capacità di stimolare la secrezione di GH. Tale attività ਠmediata dall'attivazione del recettore GHS-R 1a. Questo recettore ਠespresso principalmente a livello dell'ipotalamo e dell'ipofisi, ma ਠpresente anche in altre aree del SNC quali l'ippocampo, la pars compacta della substantia nigra, la corteccia piriforme ecc. (2) oltre ad essere presente in vari organi periferici quali lo stomaco, l'intestino, il pancreas, la tiroide, i surreni, le gonadi, il cuore ecc. Prima della scoperta della ghrelina, questo recettore orfano, si era stato visto essere specifico di una famiglia di molecole di sintesi, GHS (GH secretagoghe) sia peptidiche che non. Oltre a stimolare la secrezione di GH, la ghrelina possiede innumerevoli attività sia centrali che periferiche, tra cui: 1) stimola la secrezione di prolattina e ACTH; 2) influenza negativamente l'asse ipofisi-gonadi sia a livello centrale che periferico; 3) stimola l'appetito; 4) influenza il sonno ed il comportamento; 5) controlla la motilità gastrica e la secrezione acida; 6) modula la funzione esocrina ed endocrina del pancreas ecc. (Fig. 1). Le azioni endocrine della ghrelina necessitano dei cambiamenti post-trascrizionali che consistono nell'acilazione di una serina da parte di un acido n-octanoico o di un altro acido grasso a media catena (3). Normalmente la ghrelina non acilata ਠpresente in maggiori quantità in circolo, ma l'ormone, in tale forma chimica, non possiede attività endocrina pur mantenendo effetti cardiovascolari e antiproliferativi (4). Fig. 1 Attività biologiche della ghrelina (Aart J. Van der Lely et al. Endocrine Reviews, June 2004, 25 (3):426-457)
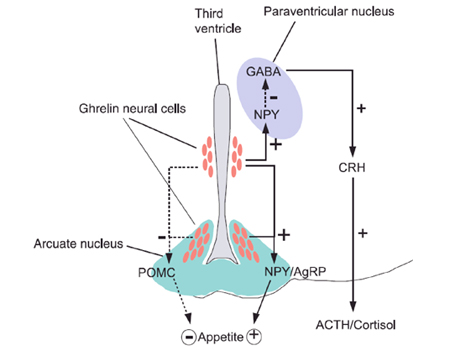
La secrezione di ghrelina ਠprincipalmente regolata da segnali metabolici e, a sua volta, l'azione di modulazione esercitata dalla ghrelina, in relazione al controllo dell'assunzione di cibo e del metabolismo energetico, sembra essere tra le sue pi๠importanti funzioni biologiche. Funzioni fisiologiche della grelina Regolazione dell'appetito L'appetito ਠcontrollato da complessi meccanismi sia a livello del SNC, in particolar modo dall'ipotalamo, sia da sostanze provenienti dai tessuti periferici. La leptina, ad esempio, prodotta dal tessuto adiposo, ਠun importante fattore di soppressione dell'appetito che trasmette un segnale di sazietà al SNC (5). I neuroni produttori di ghrelina nel nucleo arcuato (ARC) dell'ipotalamo inducono i neuroni produttori di NPY a rilasciare tale ormone, che agendo da potente neuropeptide oressizzante, stimola l'intake di cibo. La sua produzione aumenta prima dei pasti per poi ridursi circa un'ora dopo il pasto. A livello del nucleo arcuato perಠi neuroni produttori di ghrelina stimolano anche il rilascio di GABA, che, in maniera post-sinaptica modula il rilascio di pro-opio-melano-cortina (POMC), ormone anoressizzante. Sempre a livello ipotalamico ma nel nucleo paraventricolare (PVN), la ghrelina stimola il rilascio di NPY, che sopprime il rilascio di GABA, con stimolazione dei neuroni produttori di CRH, che in ultima analisi conduce ad un rilascio di ACTH e cortisolo (Fig. 2) (6). Diversi studi, negli ultimi anni hanno dimostrato che, nonostante l'ipotalamo sia la regione del SNC a pi๠alta concentrazione di GHR-S, tuttavia altre aree cerebrali subiscono l'azione della ghrelina. Fig. 2 Network ipotalamico della ghrelina. (Masayasu K et al)
Tra queste l'area tegmentale ventrale del mesencefalo che contiene una popolazione di neuroni dopaminergici che proiettano verso regioni quali l'amigdala, la corteccia prefrontale e l'ippocampo. Questa via viene comunemente definita come il sistema mesolitico dopaminergico che, si pensa, si sia evoluto come sistema di base per i comportamenti diretti alla ricerca del cibo, delle interazioni sociali e dell'accoppiamento (7). In conclusione la ghrelina ਠsignificativamente coinvolta in queste reti neuroendocrina che regolano il bilancio energetico in almeno due modi: 1) come un ormone periferico secreto dallo stomaco che, insieme ad altri segnali, come l'insulina o leptina, informa il SNC della riduzione delle riserve energetiche al fine di aumentare lo stimolo oressigenico e ridurre il dispendio energetico 2) come un neuropeptide ipotalamico che potrebbe svolgere un ruolo complementare nella regolazione dell'omeostasi energetica. Effetti pleiotropici della ghrelina Sembra molto restrittivo attribuire alla ghrelina solo un effetto oressizzante. In effetti le sue azioni sono molto pi๠numerose e complesse. Ad esempio recentemente si ਠosservato come la ghrelina promuova il sonno ad onde lente nell'uomo (8) e provochi, se iniettata nell'ippocampo, amigdala o nel nucleo dorsale del rafe dei topi, un aumento della capacità mnemonica. A livello pancreatico la somministrazione acuta di ghrelina inibisce la spontanea secrezione di insulina e quella arginina indotta, senza pregiudicare la risposta insulinica al test orale di tolleranza al glucosio (9). Inoltre la somministrazione di ghrelina nell'uomo provoca iperglicemia che non ਠdovuta al rilascio di glucagone ma probabilmente all'attivazione della glicogeno lisi o attraverso l'azione delle catecolamine o per azione diretta sugli epatociti, modulando la gluconeogenesi (10). Indagini immunoistochimiche hanno evidenziato la presenza di ghrelina anche a livello delle gonadi maschili (cellule di Leyding), ed in quelle femminili (cellule interstiziali dell'ilo ovarico), oltre ad essere stata isolata anche a livello tiroideo. In tali sedi perà², il ruolo che l'ormone potrebbe svolgere rimane ancora oscuro (11). Gli effetti cardiovascolari della ghrelina sono molteplici e si esplicano sia a livello cardiaco che dei vasi periferici. La ghrelina acilata (forma attiva) migliora la performance cardiaca sia nei soggetti sani che in pazienti con cardiomiopatia dilatativa (12). Nagaya et al hanno condotto uno studio su soggetti sani a cui veniva somministrato un bolo di ghrelina endovena. Gli effetti registrati sono stati sia una riduzione dei valori di pressione arteriosa che un aumento dell'output cardiaco (13). I livelli di ghrelina sono aumentati in alcune condizioni morbose quali l'anoressia nervosa, la cachessia cardiaca e tumorale, la Sindrome di Prader-Willy, il diabete mellito di tipo I o ancora il ghrelinoma (carcinoide intestinale). Al contrario, i livelli ematici di ghrelina risultano ridotti in condizioni quali l'obesità , la gastrectomia totale, il by-pass gastrico, l'ipertiroidismo e la sindrome di Cushing. E' interessante notare come in tutte le forme di obesità i livelli di ghrelina risultino ridotti ad esclusione della sindrome di Prader-Willy, sindrome genetica pure caratterizzata da obesità associata a ipotonia alla nascita, facies caratteristica, disturbi comportamentali ecc. (14). In un recente studio condotto su 40 bambini dai 2 mesi ai 17 anni affetti da Sindrome di Prader-Willy si ਠosservato come i livelli plasmatici di ghrelina risultavano essere aumentati in ogni fascia di età , anche durante il primo anno di vita, e comunque anche prima che questi soggetti sviluppassero obesità (15). In conclusione l'aumentata comprensione dei meccanismi d'azione nella sua interezza e complessità potranno aprire le porte sia alla comprensione dei complessi meccanismi fisiologici alla base della regolazione dell'appetito (e non solo), sia a nuove possibilità di utilizzo della ghrelina come agente terapeutico in diverse condizioni patologiche. Bibliografia 1) Date Y, Kojima M, Hosoda H et al (2000). Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141: 4255-4261. 2) Michael A. Cowley et al. The Distribution and Mechanism of Action of Ghrelin in the CNS Demonstrates a Novel Hypothalamic Circuit Regulating Energy Homeostasis. Neuron, Vol. 37, 649-661, February 20, 2003. 3) M. Gil-Campos et al. Ghrelin: a hormone regulating food intake and energy homeostasis. British Journal of Nutrition (2006), 96, 201-226 4) Cassoni P, Papotti M, Ghe C, Catapano F, Sapino A, Graziani A, Deghenghi R, Reissmann T, Ghigo E, Muccioli G 2001 Identification, characterization, and biological activity of specific receptors for natural (ghrelin) and synthetic growth hormone secretagogues and analogs in human breast carcinomas and cell lines. J Clin Endocrinol Metab 86:1738-1745. 5) Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev 60: S1-S14, 2002. 6) Masayasu Kojima, Kenji Kangawa. Ghrelin: Structure and Function. Physiol Rev 85: 495-522, 2005. 7) Alfonso Abizaid. Ghrelin and Dopamine: New Insights on the Peripheral Regulation of Appetite. Journal of Neuroendocrinology, 21, 787-793, 2009. 8) Weikel, J.C., Wichniak, A., Ising, M., Brunner, H., Friess, E., Held, K., Mathias, S., Schmid, D.A., Uhr, M. & Steiger, A. (2003) Ghrelin promotes slow-wave sleep in humans. American Journal of Physiological. Endocrinology and Metabolism, 284, E407-E415. 9) Broglio, F., Gottero, C., Benso, A., Prodam, F., Destefanis, S., Gauna, C., Maccario, M., Deghenghi, R., van der Lely, A.J. & Ghigo, E. (2003d) Effects of ghrelin on the insulin and glycemic responses to glucose, arginine, or free fatty acids load in humans. Journal of Clinical Endocrinology and Metabolism, 88, 4268-4272. 10) Broglio, F., Benso, A., Castiglioni, C., Gottero, C., Prodam, F., Destefanis, S., Gauna, C., van der Lely, A.J., Deghenghi, R., Bo, M., Arvat, E. & Ghigo, E. (2003a) The endocrine response to ghrelin as a function of gender in humans in young and elderly subjects. Journal of Clinical Endocrinology and Metabolism, 88, 1537-1542. 11) Gnanapavan, S., Kola, B., Bustin, S.A., Morris, D.G., McGee, P., Fairclough, P., Bhattacharya, S., Carpenter, R., Grossman, A.B. & Korbonits, M. (2002) The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. Journal of Clinical Endocrinology and Metabolism, 87, 2988. 12) Enomoto, M., Nagaya, N., Uematsu, M., Okumura, H., Nakagawa, E., Ono, F., Hosoda, H., Oya, H., Kojima, M., Kanmatsuse, K. & Kangawa, K. (2003) Cardiovascular and hormonal effects of subcutaneous administration of ghrelin, a novel growth hormonereleasing peptide, in healthy humans. Clinical Science (London), 105, 431-435. 13) Nagaya N, Kojima M, Uematsu M, Yamagishi M, Hosoda H, Oya H, Hayashi Y, Kangawa K. Hemodynamic and hormonal effects of human ghrelin in healthy volunteers. Am J Physiol Regul Integr Comp Physiol 280: R1483-R1487, 2001. 14) Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS 2002 Elevated plasma ghrelin levels in Prader-Willi syndrome. Nat Med 8:643-644. 15) Eva Feigerlova et al. Hyperghrelinemia Precedes Obesity in Prader-Willi Syndrome. J Clin Endocrinol Metab. July 2008, 93 (7) :2800-2805
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Immunodeficienza comune variabile Common variable immunodeficiency
Annamaria Salpietro, Valeria Ferraà¹, Antonella Talenti, Andreea Deak, Barbara Russo, Piera Vicchio, Laura Colavita, Caterina Cuppari, Silvana Briuglia, Romina Gallizzi Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina
Abstract Common variable immunodeficiency (CVID) is a primary immunodeficiency that is characterized by low level of serum immunoglobulins and an increased susceptibility to infections. The symptoms generally manifest in adults, but can occur at any age, even in infancy. Recurrent bacterial infections or pneumonias are frequent, and may be complicated by gastrointestinal problems, granulomas, autoimmune disorders. The most common autoimmune conditions are immune thrombocytopenic purpura and hemolytic anemia, pernicious anemia and systemic lupus. Mutations in the genes encoding the tumour necrosis factor (TNF) superfamily receptors transmembrane activator and calcium-modulating ligand interactor (TACI) and B cell activation factor of the TNF family receptor (BAFF-R), CD19 and the co-stimulatory molecule inducible co-stimulator molecule (ICOS) all lead to CVID and illustrate the complex interplay required to co-ordinate an effective humoral immune response. Together these defects account for perhaps 10-15% of all cases of CVID and it is highly likely that further genetic defects will be identified. Riassunto L'immunodeficienza comune variabile ਠun immunodeficit primitivo caratterizzato da bassi livelli di immunoglobuline nel siero e da un'aumentata suscettibilità alle infezioni. La sintomatologia puಠcomparire a qualsiasi età , sia nell'infanzia che in età adulta. Frequenti sono le infezioni batteriche ricorrenti e le polmoniti, oltre ad un elevata incidenza di disturbi gastrointestinali, malattie autoimmuni e insorgenza di granulomi. Le patologie autoimmuni pi๠frequentemente associate sono la porpora trombocitopenica, l'anemia emolitica autoimmune, l'anemia perniciosa, il LES ecc. Molte mutazioni a carico di diversi geni sono state associate alla malattia, tra cui il gene che codifica per il TNF, il TACI, il BAFF-R, ICOS e CD19. Nell'insieme, questi difetti, spiegano il 10-15% di casi di CVID, ed ਠaltamente probabile che ulteriori difetti genetici verranno in futuro individuati. Introduzione L'immunodeficienza comune variabile (CVID) ਠun'immunodeficienza primaria caratterizzata da un basso livello di immunoglobule sieriche ed un incremento della suscettibilità alle infezioni (1). Questi pazienti mostrano una varietà di sintomi, difetti cellulari ed immunologici. La sintomatologia insorge in genere in età adulta (seconda decade di vita) ma puಠanche insorgere nella prima infanzia. Questi pazienti sviluppano una maggiore suscettibilità alle infezioni batteriche, alle polmoniti, allo sviluppo di patologie gastrointestinali e granulomi (2-3). Questa immunodeficienza puಠanche portare ad un'aumentata predisposizione allo sviluppo di neoplasie, in particolare linfomi e malattie autoimmuni (4). L'identificazione di 4 difetti genetici che risultano nel fenotipo dell'immunodeficienza comune variabile mostra che la base genetica della malattia ਠmolto variabile (5-6-7). Dal 2003 sono state studiate le mutazioni di 4 geni nella CVID: ICOS, TNFRSF13B (TACI), TNFRSF13C (BAFF-R) e CD19. Le mutazioni in eterozigosi in TNFRSF-3B sono anche associate alla CVID, gli altri tre geni sono puramente recessivi (8-9). I recenti studi di linkage hanno identificato loci per geni CVID dominanti sui cromosomi 4q 5p e 16 q (10). La prevalenza della malattia ਠcompresa tra 1/10.000 e 1/20.000 (11). Patogenesi Il difetto che porta alla CVID consiste nel deficit maturativo della fase finale dei linfociti B. Il risultato ਠun difetto della funzione regolatoria delle cellule T con o senza la deficienza dei linfociti B. Il deficit di IL-2, IL-4 IL-5 e INF- γ dovrebbe essere associata. Gli studi genetici e molecolari hanno mostrato nelle stesse famiglie, e perfino negli stessi individui con CVID, il deficit selettivo di IgA. E' ritenuto che i fattori scatenanti di queste mutazioni che dipendono da fattori esogeni o genetici sviluppano un deficit isolato di IgA in alcuni casi e CVID in altri. Alcune di queste famiglie presentano mutazioni del gene del sistema dell' HLA-3, dei fattori C2 e C4 o TNF (12). Alterazioni immunologiche I pazienti con CVID di solito presentano ipogammaglobulinemia. La presenza di immunoglobuline normali non esclude comunque la CVID e la diagnosi definitiva richiede la conferma della mancanza della risposta anticorpale specifica (13-14-15-16). Il numero dei linfociti B ਠdi solito normale o quasi normale. Sono state trovate anche cellule B immature o anomale e questo rischio aumenta con l'età del paziente (17-18-19-20). Il maggior numero di anomalie riscontrate riguarda le alterazioni delle cellule B della memoria (CD 19+ CD 27 * IgD) ; ciಠci permette anche di classificare le differenti forme di CVID e di predire il corso del disordine in ciascun paziente (21-22-23-24-25). La riduzione delle cellule B della memoria (CD19 + CD27 + IgD) ਠassociata infatti sia negli adulti che nei bambini alle forme pi๠severe con splenomegalie e bronchiectasie (26). I pazienti con serie complicanze cliniche tendono inoltre a presentare un basso rapporto CD4/CD8. Altre anomalie immunologiche descritte consistono in: difetti dell'immunità innata e difetti dell'attivazione, dello sviluppo e della funzione delle cellule dendridiche di origine monocitica. In alcuni casi il deficit ਠaccompagnato da alterazione della produzione di IL-12 con secondaria anomalia di attivazione delle T cellule (27-28). Appare essere rilevante anche un difetto della sintesi della IL-17 soprattutto in pazienti con gravi complicanze. In questi pazienti ਠda annotare la presenza di splenomegalia, disordini autoimmuni e e aumento in circolo di linfociti CD8+ . Infine l'ultimo recente difetto ਠstato identificato sul TLR9 (toll-like receptor 9), ma ulteriori studi devono essere effettuati a tal proposito (29-30-31). Fig 1 Meccanismi di overlaping che coinvolgono l'autoimmunità della CVID
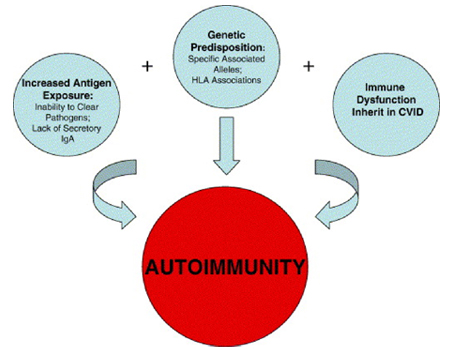
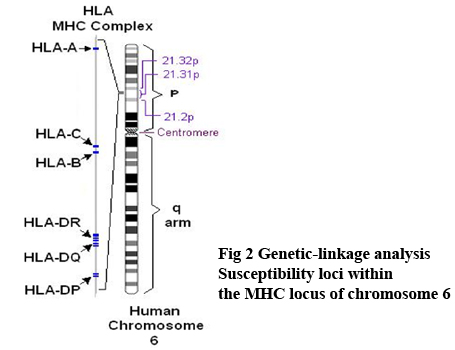
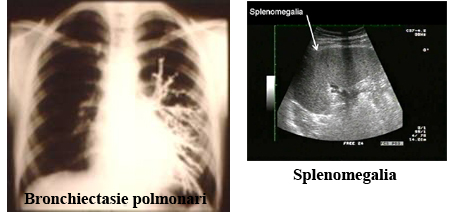
Genetica La base genetica della CVID ਠmolto variabile. Nella maggior parte dei casi la malattia si presenta in forma sporadica. Circa il 10-20 % delle forme ha un pattern familiare (32). La maggior parte delle forme familiari di CVID mostra un pattern autosomico dominante anche se un'eredità autosomica recessiva ਠpresente in una minoranza di casi. In un grande numero di famiglie l'immunodeficienza comune variabile coesiste con il deficit di IgA e un numero di pazienti affetti da deficit di IgA possono sviluppare un fenotipo CVID. Questo suggerisce che le due condizioni hanno la stessa eziologia genetica. Gli studi genetici che hanno analizzato i pazienti con deficit di IgA e CVID hanno identificato l'esistenza di un locus di suscettibilità nella regione MHC del cromosoma 6 chiamato IGAD 1 (33-34-35-36). L'eterogenicità della base genetica della CVID ਠdimostrata da recenti studi che hanno identificato loci di suscettibilita' a livello di 16q e 4 q in un numero di differenti famiglie. Fig 2 Genetic-linkage analysis;Susceptibility loci within the MHC locus of chromosome 6
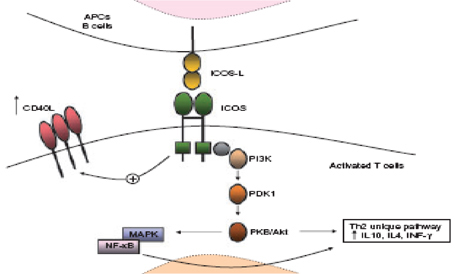
DEFICIT ICOS (INDUCIBLE CO-STIMULATOR MOLECULE) ICOS ਠespresso sui linfociti T attivati ed ਠcoinvolto nello sviluppo di citochine (IL-4, IL-5, IL-6), TNFα, IFNγ e GM-CSF ma in particolar modo nella superiduzione dell'IL-10 che provvede alla differenzazione terminale dei linfociti B a cellule della memoria e plasmacellule (37-38-39). Il deficit ICOS nell'uomo ਠstato per la prima volta descritto da Grimbacher et all. Sono stati analizzati quattro pazienti con diagnosi di CVID, con ipogammaglobulinemia con decremento di tutte le classi Ig e ricorrenti infezioni batteriche (40-41). In questi pazienti il numero delle B cellule era ridotto con decremento delle CD27IgM+IgD+. Il numero di linfociti B della memoria era diminuito. Il fenotipo e la funzione dei linfciti CD4+ era normale ma la secrezione dell'IL-10 e IL-17 era sregolata. In questi pazienti ਠstata trovata una delezione in omozigosi dell'esone 2 e 3 del gene ICOS. I dati ottenuti confermano il ruolo del gene ICOS nelle cellule T attivate e nella regolazione della differenziazione delle cellule B, nella formazione di cellule B della memoria e nella produzione finale di immunoglobuline. In totale fino ad ora in nove pazienti provenienti da quattro famiglie sono state identificate mutazioni nel gene ICOS e tutti hanno la stessa delezione (42, 43). Fig 3 Deficit ICOS
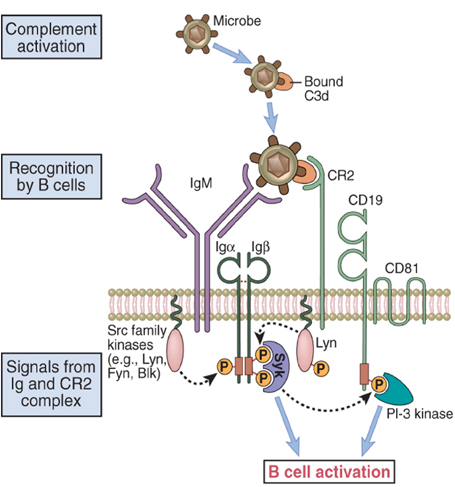
DEFICIT CD19 Lo sviluppo e la differenzazione delle B cellule ਠdipendente dal segnale di trasduzione del BCR (recettore antigene cellulare). Il co-recettore associato col BCR puಠmodulare il segnale di trasduzione del BCR positivamente o negativamente influenzando le cellule B nei vari stadi di sviluppo. La molecola CD19, insieme a CD21, CD81 e CD225 forma un complesso co-recettoriale per il BCR (44-45). CD19 ha due domini extracellulari Ig-like e una parte citoplasmatica con la capacià di legare Lyn e PI 3 Chinasi (PI3K). Dati recenti sottolineano l'importanza della PI3K. La mutazione CD19 ਠstata individuata in quattro pazienti appartenenti a due famiglie.Questi quattro pazienti mostravano infezioni batteriche ricorrenti durante l'infanzia (46-47-48). In un paziente, la sequenza del gene CD19 mostrava l'inserzione di una singola base pari (bp) CG che causava un frameshift e un prematuro codone di stop nella regione prossimale del dominio intracellulare. Negli altri tre pazienti ਠstata riscontrata una delezione di 2 bp che causava una mutazione frameshift e un prematuro codone stop nel dominio intracellulare. Il dato importante ਠche entrambe le mutazioni interferiscono sulla trasduzione del segnale di CD19. L'espressione della proteina CD19 intracellulare era infatti assente o diminuita. Il decremento di CD19 porta ad una concomitante riduzione dei livelli di CD21 mentre i membri del restante complesso recettoriale sono normali (CD81 e CD225). La conseguenza del deficit CD19 consiste in una debole risposta agli stimoli antigenici ed in una inabilità alla risposta umorale (49-50). Fig 4 Deficit CD 19
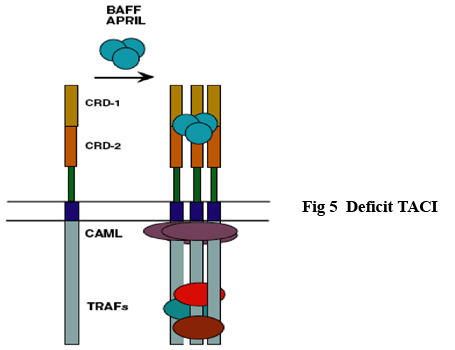
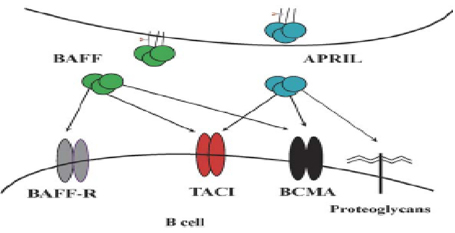
DEFICIT TACI I membri della superfamiglia del recettore del fattore di necrosi tumorale (TNFRSF) giocano un ruolo importante nella regolazione e nell'apoptosi di specifiche cellule del sistema immunitario. CD40 (TNFRSF5) ha un importante ruolo nella proliferazione e differenzazione delle cellule B e nello switching delle diverse immunoglobuline tanto che, una mutazione a livello di CD40 ਠresponsabile della Sindrome di IgM (51-52-53-54-55). Appare pertanto evidente come la funzione e la maturazione delle B cellule dipende dal corretto funzionamento dei recettori del TNF (TNFRSF) (56-57). TACI (attivatore trans membrana e modulatore del calcio e legante la ciclofilina) e BCMA (antigene di maturazione delle B cellule; TNFRSF17) sono entrambi espresse sulle B cellule e interagiscono con BAFF (attivatore B cellule della famiglia dei recettori TNF) e APRIL (ligando inducente la proliferazione) (58-59-60-61). I dati accumulati in uno studio multicentrico dall'analisi di una coorte di oltre 500 pazienti con CVID suggerisce che la mutazione in TACI ਠstata riscontrata nell'8-10 % dei pazienti (62-63). La mutazione pi๠comune consiste nella mutazione missense C104R e A181E. Nei modelli murini, inoltre, il deficit TACI si correla ad una maggiore tendenza al linfoma e ai processi autoimmunitari (64-65). Fig 5 Deficit TACI
DEFICIT BAFF-R Dall'analisi della larga coorte di pazienti precedentemente nominata ਠstato riscontrato il deficit BAFF-R solo in un paziente. Si tratta di un uomo di 60 anni con ipogammaglobulinemia. Questo soggetto presenta una delezione in omozigosi nell'esone 2 che codifica per la regione transmembrana del recettore (66-67). TACI e BAFF- R possiedono un ruolo importante nell'omeostasi delle B cellule e nello switch nelle differenti classi. Il deficit CD19 evidenzia l'importanza del segnale recettoriale dell'antigene mentre il deficit ICOS illustra l'assoluta necessità dell'interazione T-B nella risposta umorale (68). Fig 6 Deficit BAFF-R
Manifestazioni cliniche Le manifestazioni cliniche dell'Immunodeficienza comune variabile esordiscono solitamente nella seconda decade di vita. L'età di esordio dei sintomi puಠessere anche pi๠precoce (comunque oltre il secondo anno di vita) o molto pi๠tardivo (in età adulta) (69-70). Le manifestazioni cliniche in genere iniziano sotto forma di infezioni respiratorie di natura batterica complicate anni dopo da iperplasia linfoide, granulomi, processi autoimmuni, linfomi. In genere la malattia ਠdiagnosticata molti anni dopo l'inizio dei sintomi, quando sono già sopraggiunte le complicanze (71). La revisione dei dati della pi๠importante casistica di pazienti affetti da CVID dimostra che la diagnosi viene posta con un ritardo medio di 5-6 anni dall'esordio clinico. In alcuni pazienti, in cui la diagnosi ਠstata formulata solo dopo una lunga storia clinica di infezioni batteriche recidivanti, già alla prima osservazione clinica ਠpossibile dimostrare esiti permanenti quali la presenza di bronchiectasie, un quadro di broncopneumopatia cronica fino a quadri di insufficienza respiratoria, o di malassorbimento che rendono pi๠difficile il successivo controllo clinico e terapeutico (73). I sintomi di esordio sono nella maggior parte dei pazienti dovuti alla deficitaria risposta anticorpale nei confronti di piogeni e caratterizzati da infezioni batteriche recidivanti delle vie respiratorie e dell'apparato gastrointestinale. In altri pazienti, l'esordio clinico puಠessere pi๠atipico con manifestazioni cliniche spesso associate alla CVID, quali la presenza di splenomegalia, linfoadenopatia, presenza di granulomi non caseosi, malassorbimento con perdita di peso e diarrea, malattie croniche infiammatorie dell'intestino o patologie autoimmuni (anemia perniciosa, anemia emolitica, trombocitopenia, neutropenia) (74).
Infezioni Le prime manifestazioni della malattia consistono, nella maggior parte dei casi, in polmoniti e sinusiti sostenute da agenti infettivi di natura batterica. I pi๠frequenti patogeni isolati sono: Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis e differenti stafilococci. E' anche possibile riscontrare infezioni sostenute da Pneumocystis carinii, Mycoplasma pneumoniae e infezioni da Micobatteri tipici ed atipici. Anche le manifestazioni del tratto gastrointestinale sono molto frequenti (75-76-77). Tabella 1. Complicanze infettive e loro frequenza in una casistica di 248 pazienti con CVID
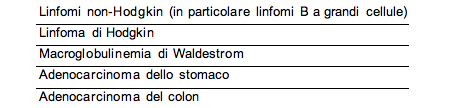
Neoplasie Un aumentato rischio di sviluppare neoplasie ਠriportata in diversi studi, anche se un calcolo esatto del rischio relativo non ਠancora disponibile. L'aumento del rischio relativo di sviluppo di linfoma varia tra il 23 ed il 100% e quello di carcinoma gastrico ਠdi circa il 50%. La tabella riporta l'elenco delle neoplasie pi๠frequentemente osservate in pazienti con CVID (78-79).
Granulomatosi L'eziologia della granulomatosi non ਠdel tutto chiara. E' stata associata con infezioni croniche dell'Herpes virus 8. Le regioni dove pi๠comunemente sono presenti i granulomi sono: i polmoni, la pelle, l'intestino o il fegato. La presenza di granulomi ਠun indice prognostico sfavorevole, essa ਠinfatti associata a malattie autoimmuni e processi linfoproliferativi (80-81-82-83). Manifestazioni gastrointestinali Molti pazienti con CVID sviluppano le malattie infiammatorie croniche intestinali RCU o morbo di Crohn (84-85). Altre condizioni patologiche riscontrabili sono: malassorbimento cronico con steatorrea e deficit di vitamina B12, enteropatia protido-disperdente, intolleranza al lattosio, atrofia dei villi e parassitosi intestinale. Sono da annotare alcune infezioni associate per lo pi๠a virus (86-87). Le infezioni gastrointestinali sono soprattutto sostenute da Salmonella, Shigella, e Campylobacter. Sono state riportate infezioni da Helicobacter Pylori nell' 80 % di pazienti con CVID e dispepsia (88). Immunodeficienza comune variabile e malattie autoimmuni Il 20% circa dei pazienti con CVID svilupperanno malattia autoimmune. In alcuni casi le manifestazioni autoimmuni possono essere il sintomo di esordio della malattia (89). La pi๠comune delle manifestazioni autoimmuni ਠla porpora trombocitopenica idiopatica e l'anemia emolitica autoimmune. Si associa spesso anche linfopenia e neutropenia autoimmune. La citopenia autoimmune ਠstata associata ad un'età precoce di insorgenza della CVID (90). E' stato dimostrato in uno studio che la citopenia insorge in 7 su 8 bambini con CVID. Sei di questi pazienti hanno presentato la sintomatologia della CVID sin dai primi anni di vita. Ad ogni modo un altro importante studio non ha mostrato una cosଠgrande incidenza di citopenia autoimmune in una popolazione pediatrica affetta da CVID (91). Nei pazienti con CVID ਠanche frequente l'interessamento gastrointestinale. I sintomi pi๠comuni sono il malassorbimento e la diarrea. La biopsia gastrointestinale mostra reperti di malattie infiammatorie croniche intestinali, malattia granulomatosa, iperplasia linfoide, atrofia intestinale sprue-like. Circa il 6 % dei pazienti con CVID ਠaffetto da malattia infiammatoria intestinale. Circa il 5- 10 % dei pazienti con CVID ਠaffetto da granulomatosi con una maggiore rappresentazione tra le popolazioni degli Ispanici e degli Afroamericani. In molti casi la malattia granulomatosa ਠstata diagnosticata prima della CVID. La splenomegalia con o senza granulomi ਠuna manifestazione comune. I pazienti con CVID e granulomi hanno un'alta incidenza di anomalie ematologiche (92). Gli altri disordini autoimmuni includono artrite idiopatica giovanile, anticorpi anti-IgA, anemia perniciosa con un'incidenza tra l'1% e il 10 %. Il LES ਠstato riportato in coincidenza con la CVID sotto l'1% dei pazienti. Ad ogni modo la relazione appare pi๠complessa. Le altre malattie autoimmune che sembrano sopraggiungere con una certa frequenza nella CVID sono: la cirrosi biliare primitiva, la tiroidite autoimmune, le vasculiti, la sindrome sicca, la vitiligine e l'alopecia (93). Tab. 2 Malattie autoimmuni e CVID
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Janus chinasi: ruolo nelle immunodeficienze e nelle neoplasie ematologiche e prospettive terapeutiche dei Jak- inibitori Janus kinase: role in immunodeficiencies and hematologic malignancies and therapeutic prospects of Jak inhibitors
Vincenzo Salpietro1, Emanuele Santantonio2, Laura Colavita1, Basilia Piraino 1, Caterina Cuppari 1, Romina Gallizzi1 1Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina 2UOC Ematologia, Università degli studi di Messina
Abstract The Janus kinase (JAK) are a family of tyrosine kinases including JAK1, JAK2, JAK3 and TYK2, all tyrosine kinase receptor involved in signal transduction mediated by numerous cytokines, involved in various capacities in the maturation and differentiation of hematopoietic cells and in regulating the immune system. The defect of Janus kinases can therefore lead to blood diseases but also to immune deficiencies in different ways depending on the affected protein and the genetic defect in question. Regarding hematologic malignancies (which we discuss the molecular and not the clinical aspects) such as we know matter most to adulthood but also in the pediatric age when can be acute lymphoblastic leukemia by mutation of JAK2. Immunodeficiencies instead are typical of pediatric age. They are the autosomal recessive SCID by a failure of function of JAK3 and Hyper-IgE syndrome from alteration of Tyk2. Finally we will mention the exciting new prospects for therapy with Jak inhibitor, immunosuppressive drugs currently on a trial that in the future could be an important tool in the treatment of many autoimmune diseases and also anti-rejection drugs. Riassunto Le Janus chinasi (JAK) sono una famiglia di tirosin-chinasi comprendente JAK1, JAK2, JAK3 E TYK2, tutte tirosin-chinasi non recettoriali coinvolte nella trasduzione del segnale mediato da numerose citochine, implicate a vario titolo nella maturazione e differenziazione delle cellule ematopoietiche e nella regolazione del sistema immunitario. Il difetto delle Janus chinasi puಠportare perciಠa malattie ematologiche ma anche ad immunodeficienze in modo variabile a seconda della proteina interessata e del difetto genetico in questione. Per quanto riguarda le neoplasie ematologiche (delle quali tratteremo l'aspetto molecolare e non clinico) queste com'ਠnoto interessano maggiormente l'età adulta e talvolta la pediatrica come nel caso della leucemia linfoblastica acuta (LAL) da mutazione del Jak2. Le immunodeficienze invece si presentano sin dall'età pediatrica e sono la SCID autosomica recessiva da difetto di funzione del Jak3 e la sindrome da iperIgE da alterazione del Tyk2. In ultimo accenneremo alle nuove esaltanti prospettive della terapia con Jak-inibitori, farmaci immunosoppressivi attualmente in via sperimentale che nel futuro potrebbero rappresentare un importantissimo strumento nella terapia di molte malattie autoimmuni e nella prevenzione dei rigetti dei trapianti. Introduzione La famiglia delle chinasi Janus ਠstata oggetto di fervido studio negli ultimi anni, e l'interesse verso questa particolare famiglia di tirosin-chinasi non recettoriali si ਠampliato fino a comprendere campi piuttosto distanti fra di loro: ਠormai stato chiarito, infatti, come queste molecole siano coinvolte nella trasduzione del segnale mediato da numerose citochine, implicate a vario titolo nella maturazione e nella differenziazione delle cellule ematopoietiche e nella generazione e regolazione delle risposte immunitarie, ed ਠstato altrettanto ben documentato il modo in cui le loro alterazioni (sia in termini di aumentata o diminuita espressione genica, sia in termini di mutazioni) contribuiscano alla patogenesi di alcune immunodeficienza e di malattie ematologiche. La famiglia JAK consta di quattro membri, JAK1, JAK2, JAK3 e TYK2, e deve il suo nome ad una delle peculiarità della struttura terziaria di queste proteine, che, presenta due domini speculari, perciಠi ricercatori si sono ispirati nell'appellarli al dio romano bicefalo Giano (Janus) conosciuto come Giano bifronte (figura 1). Nonostante le affinità architetturali, questi due domini non hanno la stessa attività catalitica, uno ha infatti una funziona catalitica, l'altro ਠdenominato dominio pseudochinasico ed ਠessenzialmente regolatorio. Figura 1- "e;Ianus"e; Giano Bifronte
Le proteine JAK presentano sette regioni di omologia, denominate domini JH (JAK homology) (figura 2) : la porzione carbossiterminale di queste molecole comprende il dominio ad attività chinasica e quello pseudochinasico, rispettivamente JH1 e JH2, il primo dei quali ਠprovvisto di tutte le caratteristiche delle molecole ad attività tirosin-chinasica, come il residuo di acido aspartico localizzato nel catalytic loop e direttamente coinvolto nel processo di trasferimento del gruppo fosfato. La porzione amino terminale si articola nei tre domini JH5, JH6 e JH7, e presenta al suo interno una sequenza FERM tramite la quale le chinasi JAK interagiscono coi recettori citochinici; la porzione rimanente, invece, si articola nei due domini JH3 e JH4, che presentano un grado significativo di omologia coi domini SH2 (Src-homology-2). Il modello descrittivo dell'attività di queste proteine attualmente accettato attribuisce al dominio JH2 una funzione inibitoria sull'attività catalitica del dominio JH1, attraverso un'interazione intramolecolare che si instaurerebbe fra l'apice dell'activation loop presente nel dominio catalitico di JH1 e l'ansa contenente il residuo V617, localizzato nel dominio JH2. Figura 2- Struttura e suddivisione in domini delle Janus Chinasi
La superfamiglia dei recettori citochinici, con cui i componenti della famiglia JAK interagiscono ai fini della efficace trasduzione del segnale, comprende proteine integrali di membrana, ciascuna dotata di un singolo segmento transmembrana, ma sprovviste di attività enzimatica intrinseca. Attraverso la loro porzione intracitoplasmatica, tuttavia, queste proteine entrano in contatto con uno o pi๠membri della famiglia JAK, attraverso le sequenze recettoriali Box-1 e Box-2. Tale superfamiglia puಠessere suddivisa in recettori di tipo I e recettori di tipo II. A fronte di queste diversità strutturali, entrambi i tipi di recettori citochinici si attivano a seguito del legame del loro specifico ligando al dominio extracellulare della proteina, che cambia conformazione e va incontro ad attiva dimerizzazione o oligomerizzazione, a seguito delle quali quale le molecole JAKs presenti sul versante intracitoplasmatico del recettore vengono a trovarsi giustapposte e innescano la cascata intracellulare del segnale attraverso una reciproca cross-fosforilazione in corrispondenza delle sequenze YY delle loro activation loop. I recettori di tipo I comprendono recettore per l'eritropoietina (figura 3), per la trombopoietina e per l'ormone della crescita e recettori eteromerici contenenti la catena comune gamma (γc) comprendenti i recettori per l'IL-2, l'IL-4, l'IL-7, l'IL-9, l'IL-13, l'IL-15 e l'IL-21. Figura 3- Janus Chinasi e recettori citochinici di tipo I
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Aspetti immunogenetici delle infezioni delle vie genitali femminili da Clamydia Trachomatis e Trichomonas Vaginalis Immunogenetics aspects of the female genital tract infection by Clamydia trachomatis and Trichomonas vaginalis
Vincenzo Salpietro1, Federica Sancetta1, Elisabetta Colonese2, Sara Manti1, Maria Concetta Cutrupi1 , Valeria Ferraà¹1, Silvana Briuglia1 1Dipartimento di Scienze Pediatriche, UOC Genetica e Immunologia Pediatrica, Università degli studi di Messina 2 UOC Ginecologia e Ostetricia, Università degli studi di Messina
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Artriti post infettive Post-infectious arthritis
Antonella Talenti, Donatella Comito, Andreea Deak, Sara Manti, Basilia Piraino, Caterina Munafà², Caterina Cuppari, Romina Gallizzi Dipartimento di Scienze Pediatriche, UOC Genetica ed Immunologia Pediatrica, Università degli studi di Messina
Abstract In this article we reported a systematic review about post-infectious arthritis, their classification in pediatric age, management and treatment respectively, with practice guidelines. In ambit of post infectious arthritis we analysed three condition: reactive arthritis, Lyme disease and rheumatic fever. Reactive arthritis is a non-purulent joint inflammation that usually follows bacterial gastrointestinal or urogenital infections. Management can differ according to the triggering infection. Lyme disease is the most frequent tick-borne infectious disease in Europe. Borrelia B. is present in approximately 5 % to 35 % of sheep ticks. Diagnosis is made on the basis of the clinical symptoms, and in the stage II and III by detection of Borrelia-specific antibody. Acute rheumatic fever is a very common cause of cardiovascular mortality and morbidity all over the world. Diagnosis is based on Jones criteria. Eradication of Streptococcical infectious is necessary with appropriate antibiotics. Riassunto In questo articolo viene riportata una ampia illustrazione relativa alle artriti post-infettive in particolare in età pediatrica, che comprendono l'artrite reattiva, la malattia di Lyme e la malattia reumatica. L'artrite reattiva costituisce una condizione infiammatoria non-settica a carico dell'articolazione secondaria ad un processo infettivo che puಠavere una origine primaria diversa. La malattia di Lyme ਠcausata dalla puntura di un artropode che una volta inoculato l'agente patogeno, Borrelia B, determina un quadro clinico peculiare, la diagnosi soprattutto nelle fasi avanzate ਠdi tipo sierologico, il trattamento varia a seconda dello stadio clinico. La malattia reumatica, in pasato considerata una delle maggiori cause cardiovascolari di morte tra i soggetti giovani, ਠdovuta all'infezione Streptococcica e, una volta effettuata la diagnosi, ਠnecessario avviare il trattamento secondo le pi๠recenti Linee Guida. Introduzione Esistono diverse forme di artrite correlate ad infezioni: virali, batteriche, parassitarie ecc. Nell'ambito delle principali malattie reumatologiche in età pediatrica si distinguono essenzialmente artriti infettive e artriti postinfettive (Tab.1). Tra le prime l'artrite settica ਠdeterminata dall'invasione nell'articolazione dell'agente infettivo che puಠessere identificato nel liquido sinoviale; nelle artriti postinfettive l'artrite si sviluppa in una sede diversa dall'infezione primaria, che puಠessere il tratto gastrointestinale, urogenitale ecc. In tale condizione non ਠsempre possibile identificare l'agente infettivo responsabile a livello dell'articolazione coinvolta e la sintomatologia articolare puಠseguire anche di alcune settimane l'evento infettivo acuto. Tab 1 Principali malattie reumatologiche in età Pediatrica
Cassidy, 2005 Nell'ambito delle artriti post-infettive passeremo alla trattazione di tre condizioni patologiche emblematiche: artrite reattiva, malattia di Lyme e malattia reumatica. Artrite reattiva L'artrite reattiva (ReA) ha un'incidenza stimata intorno a 30-200: 100.000 nella popolazione, colpisce in genere una fascia di età compresa tra 20-40 anni e si manifesta in maniera uguale tra i sessi. Diversi fattori contribuiscono alla sua patogenesi: 1. la presenza di batteri e/o prodotti batterici a livello delle giunzioni articolari e la successiva risposta da parte del nostro sistema immunitario; 2. l'effetto di peptidi "e;artritogeni"e; derivati da alcuni ceppi batterici (Salmonella, Yersinia) e la presentazione dell'antigene da parte dei fagociti ai linfociti T citotossici a livello della membrana sinoviale; 3. la formazione di immunocomplessi IC tra gli antigeni e le molecole HLA di classe I e II, presentati ai linfociti CD4+ e CD8+ (in corso di infezione da Chlamydia) ; 4. il persistente stato di infezione batterica e lo squilibrio tra TNF alfa, INF gamma, IL-12, IL-10; Fattori genetici giocano un ruolo importante nell'eziologia delle ReA. L'antigene HLAB27, ਠstato riscontrato in circa il 65-80% dei pazienti con artrite reattiva. I pi๠comuni agenti patogeni comprendono: "e;¢ Gram Negativi (Enterobacteriaceae, Salmonella, Yersinia, Campylobacter, Shighella) "e;¢ Chlamydia (C. Trachomatis, C Pneumoniae) ; "e;¢ Clostridium Difficile "e;¢ Vibrio Parahaemolyticus; "e;¢ Mycobacterium bovis; "e;¢ Mycobacterium Tuberculosis; "e;¢ Specie Mycoplasma (Ureoplasma urealyticum). L'artrite di solito colpisce le grandi articolazioni degli arti inferiori senza erosioni. Le ginocchia sono interessate nel 70%, le caviglie nel 57 %, le dita nel 35 %, il polso e le dita sono coinvolti nella maggior parte dei pazienti (45%) (Tab 2). La durata dell'artrite ਠdi 4-5 mesi, ma in circa la metà dei pazienti vi puಠessere un coinvolgimento muscolo-scheletrico che puಠpersistere pi๠di un anno. Circa il 15-30% dei pazienti sviluppa artriti ricorrenti o croniche, sacro ileiti, spondiliti. Molti pazienti possono avere un sierologia HLA-B27 positiva. L'infiammazione dei legamenti e dei tendini in corrispondenza dei siti di giunzione alle strutture ossee, ਠpresente nel 30 % dei casi. Tab 2
La diagnosi si avvale degli esami di laboratorio e strumentali. Esami di laboratorio: le anomalie di laboratorio sono aspecifiche, aumento della VES, della PCR, anemia normocitica normocromica, moderata neutrofilia. All'esame delle urine ਠpossibile documentare piuria asettica. Il FR e gli anticorpi antinucleari sono negativi. Test microbiologici (esami sulle feci), solo per quei paziente la cui sintomatologia sia esordita con "e;enterite"e;. Yersinia e Salmonella sono state isolate nel 9% dei pazienti che hanno presentato diarrea almeno 4 settimana prima. La diagnosi di infezione da Enterobacteriaceae si basa sui tests sierologici attraverso l'uso dei metodi ELISA. La specificità di tali metodi ਠpari al 90%. à utile dosare le immunoglobuline di classe IgA, IgG e IgM, mentre nelle forme croniche solo le IgG e IgA. Nei pazienti con Salmonella tali anticorpi possono rimanere elevati per un periodo di tempo pari a 9-14 mesi rispetto ai casi di enterocolite (4 mesi). Anche la diagnosi di artrite secondaria ad infezione da Chlamydia ਠdi tipo sierologico. à possibile dosare gli anticorpi nel siero e a livello del liquido sinoviale (LS), in quest'ultimo caso la specificità delle IgG ਠpari all'80%. Inoltre un altra tecnica in grado di confermare la diagnosi di Chlamydia ਠla PCR, la sensibilità e la specificità di tale indagine ਠdel 100% . L'HLA-B27 rappresenta un marker prognostico in caso di ReA. I pazienti sieropositivi hanno una maggiore possibilità di sviluppare artrite cronica e/o ricorrente, uveite, sacro ileite e spondilite . L'aspirazione e l'esame del LS, nella fase acuta di alcune forme di artriti, consentono di documentare l'incremento delle cellule della serie bianca, in particolare neutrofili. Le colture di liquido sinoviale sono spesso negative. Esami radiologici: 1) RX articolare : alterazioni di tipo erosivo sono state riscontrate nel 12 % dei pazienti, soprattutto a carico delle piccole articolazioni. Sono frequenti le localizzazioni a carico della colonna toraco-lombare. Ossificazione asimmetrica paravertebrale e sindesmofiti possono essere osservati a livello della faccia anteriore dei corpi vertebrali; 2) Scintigrafia con Tc 99, ਠuna tecnica dotata di sensibilità soprattutto per identificare l'entesopatia nella fase precoce di tale disordine; 3) RMN puಠessere utile per documentare il coinvolgimento delle articolazioni sacroiliache nel caso in cui l'esame radiografico fosse risultato negativo; 4) ecotomografia, ਠuna metodica di I livello in grado di dimostrare le lesioni a carico dei tessuti molli (entesite, sinovite). La diagnosi di ReA rimane un problema. Non vi sono dei criteri clinici su popolazione pediatrica, vengono, infatti utilizzati i criteri dell'ACR (American College of Rheumatology) (Tab.3). Tab 3 Criteri diagnostici ACR
La diagnosi differenziale comprende: - altre spondilo artropatie; - artrite settica; - artriti infezioni-correlate, Malattia di Lyme, artriti virali, artriti da parassiti, da Brucella, artriti acquisite (gonococciche, HIV) ; - artrite da sarcoidosi; - sindrome di Behcet. La malattia di Lyme à stata riconosciuta come entità nosologica già a partire dal 1975. L'agente patogeno la Borrelia Bugdoferi fu identificata circa 26 anni addietro. Tale organismo ਠstato a sua volta riconosciuto quale responsabile dell'eritema cronico migrante. La scoperta inoltre di tale microorganismo ha consentito l'identificazione di altri sottotipi di Borrelia (B. garini, B. afzelii, B. spielmanii). In Europa, come nel Nord America, vi sono diverse specie cliniche di Borrelia bugdoferi in grado di determinare malattia nell'uomo. Borrelia Spielmanii ਠstata descritta come agente causale dell'eritema migrante. Borrelia afelii ਠconosciuta quale patogeno di quella condizione cutanea conosciuta come "e;acrodermatite cronica atrofica"e;. Borrelia garinii ਠnota come causa di alcuni casi di neuroborreliosi. La Borrelia ਠun Gram negativo, fa parte del gruppo delle spirochete, si moltiplica lentamente e solo in terreni di coltura speciali. Presenta sulla membrana cellulare, esternamente, a livello dei flagelli, delle proteine basiche, le pi๠importanti sono Osp A, B e C, che oltre alla proteina flagellare, vengono riconosciuti come antigeni dal sistema immunitario. La trasmissione all'uomo avviene in genere attraverso un vettore quasi sempre un artropode (zecca). L'uomo rappresenta un ospite occasionale. L'infezione e le sue manifestazioni possono essere precoci o tardive. In alcuni casi l'infezione puಠdecorrere in maniera asintomatica. La probabilità di trasmissione all'uomo dopo la puntura da parte della zecca ਠbassa nelle prime 24 h dopo l'adesione alla sede di inoculo, aumenta successivamente. Solo dopo il contatto con il sangue che la Borrelia migra dall'intestino delle zecca alle ghiandole salivari e viene cosଠrilasciata. Nei bambini le zecche si localizzano pi๠di frequente a livello del cuoio capelluto. Qualsiasi tipo di manipolazione come schiacciare e/o applicare olii e altri medicamenti, dovrebbe essere evitato, tali manovre potrebbero favorire il passaggio in circolo e quindi la trasmissione dell'infezione. L'infezione da parte della Borrelia Bugdoferi puಠmanifestarsi nell'89 % dei casi con eritema migrante, 3 % neuroborreliosi, linfocitoma 2 %, cardite in meno dell'1% dei casi, artrite nel 5 % e come acrodermatite cronica atrofica nell'1 %. Manifestazioni Cliniche La malattia di Lyme puಠcolpire diversi organi e le manifestazioni cliniche possono essere distinte in locali e generalizzate. - Stadio I: (giorni o settimane dopo la puntura della zecca) eritema migrante intorno alla sede di infezione; - Stadio II: (settimane fino a 6 mesi dall'inoculo), meningoradiculite (Sindrome di Bannwarth), meningite, paralisi del faciale, encefalite, artrite, mialgia, eritema multiplo, miocardite e/o pericardite, irite; - Stadio III (pi๠di 6 mesi dopo l'inoculo), encefalite e/o enecefalomielite, arterite cerebrale, polineuropatia, mono-oligoartrite, acrodermatite cronica atrofica; Nello stadio I l'eritema migrante ਠpresente nell'80% dei casi, consiste nella presenza di una area circolare e/o ovalare intorno alla sede di infezione, che puಠandare incontro ad aumento di diametro. Se non trattato l'eritema puಠrisolversi spontaneamente in pochi giorni dall'esordio. Puಠessere associato ad altri sintomi, malessere, modesto rialzo febbrile, dolore a carico delle piccole articolazioni, astenia. Nello stadio II la meningoradiculoneurite ਠla seconda manifestazione, dopo l'eritema migrante, pi๠comune della malattia di Lyme. Si manifesta con linfomonocitosi, radicolite, deficit dei nervi cranici (paralisi bilaterale del faciale), aumento della concentrazione proteica nel CFS. Nei bambini, sono frequenti paralisi del faciale recidivante, meningite linfocitica in assenza di focolai neurologici. (Fig 1 a, b, c)
La cardite da Borrelia si manifesta nell'1% dei casi, con una mediana di 21 giorni, si puಠassociare ad altre manifestazioni come eritema migrante e deficit neurologici. I sintomi clinici comprendono vertigini, cardiopalmo, episodi sincopali, disturbi della conduzione cardiaca (blocco AV, anomalie del tratto ST e T che indicano la presenza di miocardite). Il linfocitoma da Borrelia, ਠuna condizione benigna che si manifesta pi๠comunemente nei bambini a livello dei capezzoli, scroto, naso o arti superiori negli adulti. Nello stadio III, sei mesi o diversi anni dopo la puntura della zecca, le manifestazioni cutanee dapprima di tipo infiammatorio, possono andare incontro ad atrofia. Queste si localizzano a livello della superficie estensoria degli arti, solo in maniera occasionale al volto e al tronco. Consistono in lesioni, cutanee assottigliate e secche a volte associate ad alterazioni della pigmentazione (Fig 2). Fig 2: Manifestazioni cutanee M. Lyme a) Eritema maculare migrante - b) Eritema anulare migrante - c) Linfocitoma del lobo - d) Acrodermatite cronica atrofica
Metodi Diagnostici La Borrelia puಠessere coltivata in terreni speciali, la PCR ਠin questo caso un test con una sensibilità diagnostica limitata. La diagnosi a sua volta ਠbasata sui criteri clinici e sulla storia anamnestica. Nel sospetto di malattia di Lyme ਠnecessario avviare le indagini sierologiche e in caso di esami negativi o borderline, ਠutile ripetere gli esami su siero per tre settimane. Le linee guida raccomandano la ricerca degli anticorpi IgG e IgM. Gli Ab anti Borrelia sono stati identificati in pi๠del 50% dei pazienti con eritema migrante. La sierologia potrebbe essere negativa nella fase iniziale dell'infezione o quando sia stata già avviata terapia antibiotica . In caso di neuroborreliosi, l'esame del CFS dimostra la presenza di pleiocitosi, di solito leucociti 1000/ml, dove i linfociti sembrano predominare. La PCR potrebbe essere utilizzata a completamento della diagnosi. Trattamento La terapia antibiotica se iniziata in tempi precoci ਠin grado di prevenire le complicanze e l'evoluzione verso una forma di infezione di tipo cronico, secondo le seguenti linee guida. Tab 4: Durata media raccomandata del trattamento
La Malattia Reumatica La Malattia Reumatica (MR) ਠun disordine multi sistemico secondario all'infezione da parte dello Streptococco à emolitico di gruppo A, generalmente responsabile di infezioni a carico delle alte vie aeree quali faringiti e/o faringotonsilliti. Gli aspetti clinici vengono descritti dai Criteri di Jones come segue. Tab 5: Criteri diagnostici di Jones
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Neurofibromatosi di tipo 1: aspetti genetici e clinici Neurofibromatosis type 1: genetic and clinical aspects
Giovanna Elisa Calabrà², Maria Concetta Cutrupi, Annamaria Salpietro, Valeria Ferraà¹, Caterina Munafà², Romina Gallizzi, Silvana Briuglia Dipartimento di Scienze Pediatriche, UOC di Genetica ed Immunologia Pediatrica, Università degli studi di Messina
Abstract Neurofibromatosis 1 (NF1- OMIM: 162200), formerly termed von Recklinghausen's disease, is an autosomal dominant neurocutaneous disorder with a birth incidence of one in 2500 and a minimum prevalence of one in 4-5000. A high number of cases (up to 50%) are sporadic. The NF1 gene is located on chromosome 17q11.2 and the protein product termed neurofibromin is strongly expressed in the nervous system and with the role as a negative regular of the ras proteins signal. This condition appears to be fully penetrant but has a highly variable expression, even within families. Diagnosis is based on the clinical criteria recommended by an NIH Consensus Conference which include multiple cafà¨-au-lait spots, cutaneous or subcutaneous neurofibromas (benign peripheral nerve sheath tumours), plexiform neurofibromas, axillary or inguinal freckling, optic gliomas, and iris Lisch nodules (hamartomas diagnosed on slit-lamp examination). Complications occur in some patients and include learning difficulties or mental retardation, focal neurological deficits, dysplastic skeletal lesions, hypertension, and, rarely, malignancy. NF1 is a complex neurocutaneous disease requiring supervision and management by an expert multidisciplinary team. Riassunto La NF1 (OMIM: 162200), anche detta Malattia di Von Recklinghause, ਠuna tra le pi๠comuni malattie autosomiche dominanti con una incidenza di 1 su 2500-3000 nati ed una prevalenza di circa 1 su 4000-5000 individui nella popolazione generale. Il 50% circa dei casi sono sporadici. E' una malattia ad espressività molto variabile (soprattutto interfamiliare) e penetranza quasi completa ed età dipendente (ਠcompleta oltre i 5-6 anni). Il gene malattia, di circa 60 esoni, ਠlocalizzato sul cromosoma 17 (17q11.2) e codifica per una proteina, la neurofibromina, regolatrice della crescita e differenziazione cellulare. Clinicamente, ਠcaratterizzata da tre tipi di manifestazioni: (1) segni clinici principali (che si manifestano nella grande maggioranza degli individui affetti e fanno parte dei criteri diagnostici) : macchie caffe' latte (>6), efelidi (lentigginosi) ascellare, inguinale e/o della base del collo o del tronco, noduli iridei di Lisch e neurofibromi; (2) segni clinici accessori (presenti in una discreta percentuale di individui affetti, ma non tali da entrare a far parte dei criteri diagnostici) : macrocefalia, statura al 10°-25° percentile, anomalie toraciche (pectus excavatum o carenatum), ipertelorismo; (3) complicanze (variabili e poco frequenti, ma spesso gravi ed invalidanti) : disturbi cognitivi e dell'apprendimento, neurofibroma plessiforme, scoliosi, displasia delle ossa lunghe, complicanze neurologiche, ipertensione arteriosa, malformazioni cardiovascolari. La varieta' dell'espressione clinica, il rischio di tumori e l'imprevedibilita' dell'evoluzione impongono un monitoraggio attento ed un approccio multidisciplinare dei pazienti con NF1. Introduzione La Neurofibromatosi di tipo 1 (NF1) o malattia di von Recklinghausen fa parte di un gruppo di malattie genetiche multisistemiche e progressive dette Facomatosi o anche sindromi "e;neurocutanee"e; (sclerosi tuberosa, la malattia di Sturge-Weber, la malattia di von Hippel-Lindau, l'atassia-teleangectasia, la sindrome di Jadassohn, l'ipomelanosi di Ito e l'incontinentia pigmenti) le cui manifestazioni cliniche, estremamente variabili ed eterogenee, riguardano primariamente, ma non esclusivamente, i tessuti di derivazione neuroectodermica (1). Si tratta di un gruppo di affezioni con alcune caratteristiche comuni, ma che rappresentano entità cliniche distinte la cui differenziazione ਠmolto importante in quanto la storia naturale, il follow-up e la consulenza genetica variano a seconda delle diverse forme. Oggi solo la NF1 e la neurofibromatosi tipo 2 (NF2, forma centrale o acustica) appaiono ben caratterizzate dal punto di vista clinico e distinte dal punto di vista molecolare. Una terza forma la schwannomatosi ਠben caratterizzata dal punto di vista clinico, in quanto caratterizzata dalla presenza di schwannomi multipli ma in assenza di altre manifestazioni cliniche della NF2, ma non ancora dal punto di vista genetico in quanto si ipotizza che possa essere una forma allelica della NF2 o forma il cui gene ਠlocalizzato sul cromosoma 22 in regioni diverse da quello della NF2 (2). La NF1 ਠla forma pi๠comune di Facomatosi con un'incidenza di 1 su 2500-3000 nati ed una prevalenza di circa 1 su 4000-5000 individui nella popolazione generale. Si trasmette con modalita' autosomica dominante, il 50% dei casi sono sporadici (3). E' una malattia ad espressività molto variabile, anche all'interno della stessa famiglia, e penetranza quasi completa, età dipendente (i segni clinici compaiono progressivamente fino a diventare del 100% all'età di 6-8 anni) (4). Il gene malattia (gene NF1) ਠstato localizzato in sede pericentrometrica del braccio lungo del cromosoma 17 (17q11.2) (Fig.1) , nel 1987 (5). Il gene NF1 ਠun gene oncosoppressore di oltre 335 kb di DNA genomico, con almeno 60 esoni, che codifica per una proteina di 2818 aa chiamata neurofibromina (Fig. 2) che si localizza nei microtubuli citoplasmatici; essa ha un dominio omologo alla famiglia di proteine GTPasi-attivanti (GAP), che costituisce circa il 13% dell'intera sequenza codificante e che svolge una regolazione negativa della crescita cellulare con attività di controllo di attivazione sul ras pathway. Da qui la funzione di soppressore di tumore svolto dalla neurofibromina (6). Il ras pathway (Fig. 3) sembra essere coinvolto, oltre che nella NF1, anche in altre sindromi genetiche associate ad una maggiore predisposizione all'insorgenza dei tumori, quali la sindrome di Costello, la s. di Noonan, la s. di Leopard, la s. di Legius (7). Fig. 1-2: Gene NF1 (17q11.2) e Neurofibromina
Ad oggi si conoscono oltre 450 mutazioni (delezioni, inserzioni, duplicazioni, sostituzioni nucleotidiche) a carico del gene NF1 ma senza una reale associazione genotipo-fenotipo, fatta eccezione per le ampie delezioni associate ad un fenotipo severo con ritardo mentale e dismorfismi facciali (8). Nel 2007 M. Upadhyaya et al. proposero, sull'American Journal of Human Genetics, un'altra importante correlazione genotipo-fenotipo, ossia la delezione c.2970-2972 delAAT sull'esone 17 del gene NF1 e l'assenza di neurofibromi cutanei (9). Fig. 3: Ras pathway e sindromi associate (da: Oren N. Gottfried et al. Neurosurg Focus 28 (1):E8, 2010)
Manifestazioni cliniche e complicanze della NF1 La NF1 ਠuna malattia genetica multisistemica che si presenta con segni clinici che possono interessare vari organi (cute, sistema nervoso periferico, scheletrico e cardiovascolare) e con possibili complicazioni di varia natura e gravità (vascolari, tumorali) che compaiono nel tempo. Il segno clinico patognominico della NF1 ਠrappresentato dalle macchie caffà¨-latte (Fig.4-5), primo segno della malattia, di forma variabile, di diametro fra 10 e 30 mm, a margini netti e di colore uniforme.Compaiono alla nascita o entro il 1° anno d'età e aumentano di n° e/o dimensioni fino al 5°-6° anno di vita. Sono diffuse a tutta la superficie corporea, con predilezione per tronco e arti, e risparmio di volto e regioni palmo-plantari. Sono presenti nel 95% delle persone con NF1 (10). Per costituire un criterio clinico devono essere almeno 6 e con di diametro superiore a 5 mm prima della pubertà e oltre il 15 mm in età adulta (11). Fig. 4-5: Macchie caffਠlatte in una nostra paziente affetta da NF1
Nella NF1 sono presenti altre manifestazioni cutanee quali: - lentigginosi nelle aree di frizione cutanea (ascelle e inguine) o freeckling ascellari o inguinali (Fig. 6) : aree iperpigmentate del tutto simili alle macchie caffਠlatte ma di dimensioni inferiori, di 2-3 mm di diametro, presenti alla nascita o che compaiono nei primi 6-7 anni di vita. Possono localizzarsi anche alla base del collo, sul tronco, a livello delle zone periorale e perioculare (12). Sono in genere il secondo segno che compare, dopo i 2-3 anni, e sono presenti nell'85% delle persone con NF1. Fig. 6: Freckling ascellare ed inguinale in nostre pazienti affette da NF1
- neurofibromi cutanei o sottocutanei (Fig.7-8) : piccole masserelle molli elastiche che compaiono in genere alla pubertà , raramente prima dei 7 anni o durante la gravidanza (13), il che suggerisce un'influenza ormonale. Possono essere isolati o, pi๠frequentemente multipli, in alcuni casi molto numerosi; sono tumori benigni dei nervi periferici e non interessano altri tessuti, ma possono crescere e comprimere i tessuti circostanti, come il neurofibroma spinale (14). I neurofibromi sottocutanei costituiscono un fattore di rischio di presenza di sottostante neurofibroma plessiforme o di un tumore maligno della guaina dei nervi periferici il cui acronimo inglese ਠMPNST (malignant peripheral nerve sheath tumour). I neurofibromi sono presenti nel 98% degli adulti con NF1. Per costituire un segno clinico devono essere 2 o pià¹. Fig. 7-8-9: Neurofibromi cutanei in nostre pazienti adulte affette da NF1
- noduli iridei di Lisch (Fig. 11) : amartomi asintomatici presenti sulla superficie dell'iride, visibili solo con la "e;lampada a fessura"e; come masse tridimensionali traslucide, punteggiate da cellule contenenti melanina. Compaiono in genere dopo i 5-6 anni (17). Sono presenti nel 95% delle persone con NF1. Non hanno alcun significato clinico se non diagnostico quando sono due o pià¹. Fig. 11: Noduli iridei di Lisch
- glioma delle vie ottiche: tumore benigno a bassa crescita interessante le vie ottiche (nervo e talora anche il chiasma ottico) a comparsa entro i primi 10 anni di vita (18). E' presente nel 15% dei bambini con NF1 ed individuabile solo con Risonanza magnetica o TAC cerebrale. Solo nel 2-5% dei casi puಠdare problemi oculari (diminuzione della vista, strabismo, protrusione del globo oculare), pubertà precoce o progredire, e di solito questo succede entro l'età di 6 anni, raramente dopo e comunque entro i 10 anni. - displasia scheletrica: anomalia di alcune ossa lunghe (tibia, fibula) che tendono a rompersi e a non ripararsi, dando luogo ad un aspetto radiologico tipo "e;pseudoartrosi"e;, o dell'osso sfenoide (19) Sono anomalie già presenti alla nascita ma infrequenti (5% delle persone con NF1). Vi sono poi altri segni clinici meno frequenti ma molto caratteristici della NF1, quali: disturbi di apprendimento (50% dei casi) identificabili nei primi anni di vita con difficoltà di lettura, di linguaggio, deficit di attenzione, con quoziente intellettivo in genere normale (20) ; macrocefalia sin dai primi anni di vita (40% delle persone con NF1) ; bassa statura (40%) ; scoliosi (10%) non specifica o "e;distrofica"e; (quest'ultima ਠprogressiva tra i 6 e i 10 anni) (21). Il decorso clinico della malattia non ਠprevedibile. In tre quarti dei casi la NF1 ਠrelativamente benigna, ma nel restante 25% dei casi si sviluppano una o pi๠complicazioni, alcune delle quali determinano nel complesso una riduzione dell'attesa di vita. Le principali complicanze della NF1 sono le seguenti: - Neurologiche: (glioma delle vie ottiche complicato con anomalie oculari e ipotalamiche, difficoltà di apprendimento, compressione da neurofibroma dei nervi periferici, spinali e del midollo spinale). Nei pazienti con NF1 possono essere riscontrate le cosiddette Unidentified Bright Objects (UBO), ossia piccole aree asimmetriche di forma rotondeggiante od ovale, a margini sfumati, che non causano effetto massa, localizzate in sede talamica, capsula interna, cervelletto e tronco encefalico e visibili alla RMN come lesioni iperintense in T2.Tendono a scomparire con l'età e sono del tutto asintomatiche (22) ; - Ortopediche: (displasia tibiale, dell'ala dello sfenoide con protrusione del bulbo oculare, scoliosi precoce e rilevante) (23) ; - Cardiovascolari: costituiscono, insieme con i tumori a rapida crescita, la principale causa di morte soprattutto nell'adolescenza (24) : l'ipertensione arteriosa, anche in età infantile e relativamente comune, dovuta a cause diverse (restringimenti vascolari o tumore surrenalico) ; l'emorragia cerebrale che ਠresponsabile del 50% della mortalità . Meno frequentemente (2%) vi ਠuna malformazione cardiaca, in genere una stenosi polmonare (25). - Tumori maligni (soprattutto MPNST) dovuti ad ulteriori mutazioni somatiche di altri geni, oltre al gene NF1, e che sembra si sviluppino in genere in sede di neurofibroma sottocutaneo e soprattutto plessiforme (26). La varietà dell'espressione clinica, il rischio di tumori e l'imprevedibilità dell'evoluzione clinica impone un monitoraggio regolare ed attento dei pazienti con NF1. Il genetista rappresenta il "e;regista"e; nella gestione del paziente affetto da NF1 e nella valutazione dei parenti e del loro rischio di ricorrenza, ma l'approccio al paziente con NF1 ਠsicuramente multidisciplinare e comprende un'attenta valutazione dei parametri auxologici, una valutazione dermatologica, ortopedica, neurologica e comportamentale, oculistica, audiologica ed, inoltre, ਠfondamentale il monitoraggio pressorio dato l'aumentato rischio, in questi pazienti, di sviluppare ipertensione arteriosa. Fondamentale sarà la sorveglianza per lo sviluppo di tumori e soprattutto di MPNST. A tal fine andrà prestata attenzione alla comparsa di dolore persistente o crescita improvvisa di una massa o modificazione dell'aspetto cutaneo e comparsa di segni neurologici, soprattutto per neurofibromi sottocutanei o plessiformi (soprattutto se in sede brachiale o lombare), se una sede ਠstata irradiata per una precedente terapia, nel caso di precedente tumore maligno della persona affetta o di un parente di I grado, se la mutazione causa della NF1 ਠuna microdelezione. Negli ultimi anni la ricerca si ਠdibattuta sull'eventuale indicazione ad effettuare, in follow up, anche nei pazienti asintomatici, una valutazione imaging con RMN encefalo per prevenire l'insorgenza di tumori cerebrali. In atto non sembra sussistere, in assenza di sintomatologia, l'indicazione ad effettuare di routine la RMN encefalo nei pazienti con NF1 (27). NF1: diagnosi clinica e molecolare La diagnosi di Neurofibromatosi, ad oggi, ਠessenzialmente clinica, e si basa sui criteri diagnostici internazionali NIH, pubblicati nel 1988 (28). La diagnosi di NF1 si pone in presenza di 2 o piu' dei seguenti criteri diagnostici (Tab. 1). Tab. 1: Criteri diagnostici NIH, 1988
Nel paziente adulto la diagnosi ਠgeneralmente pi๠semplice rispetto al paziente pediatrico, in quanto nel bambino spesso le macchie caffਠlatte rappresentano l'unica manifestazione clinica della malattia (29). La sintomatologia evolve con l'età e la penetranza ਠquasi completa intorno al 5 anno di vita. Le manifestazioni cutanee si presentano progressivamente. Le macchie caffਠlatte possono essere già presenti, in numero sufficiente, alla nascita ma possono anche presentarsi nel corso dei primi anni di vita. I neurofibromi cutanei sono rari in età infantile e generalmente compaiono in età preadolescenziale. I neurofibromi plessiformi possono essere presenti gia' alla nascita (30). La varietà dell'espressione clinica e l'evoluzione imprevedibile della malattia implicano un follow up attento del paziente affetto da NF1, soprattutto del paziente pediatrico in cui i segni clinici sono spesso insufficienti ed il fenotipo variabile. La diagnosi clinica, in alcuni casi, puಠessere confermata dalla diagnosi molecolare. La diagnosi molecolare serve per identificare le mutazioni a carico del gene NF1. Puಠessere utilizzata la tecnica del Test della Proteina Troncata (PTT) la cui sensibilità ਠrelativamente elevata in quanto, l'80% circa delle mutazioni NF1 sono da sfasamento del registro di lettura che comportano appunto una proteina pi๠corta. Per la sua complessità , tuttavia, il PTT viene limitato ai casi sporadici. La FISH (Fluorescent In Situ Hibrydization) viene utilizzata per le ampie delezioni (< 10%) e quando c'ਠil sospetto clinico di delezione, (fenotipo severo), anche nei casi familiari. Nei casi familiari si puಠricorrere anche all'analisi di linkage (31). In ogni caso, la diagnosi molecolare puಠessere effettuata per i casi sporadici con segni clinici dubbi: in genere bambini di età inferiore a 6 anni che presentano un solo segno diagnostico come la presenza di macchie caffà¨-latte, e per i quali la probabilità di essere affetti ਠdi circa il 60% o come diagnosi preclinica nei figli di persone affette. Negli ultimi anni ci si ਠmolto concentrati su un'altra tecnica per l'identificazione delle mutazioni NF1: la DHPLC (Denaturing HighPperformance Liquid Chromatography) (32). De Luca et al. in un articolo pubblicato su Human Mutation (2004) ha messo a confronto le due tecniche, PTT e DHPLC, in 110 pazienti con NF1, sottolineando l'efficacia della DHPLC, risultata in grado di identificare il maggior numero di mutazioni del gene NF1 rispetto la PTT (33). La diagnosi molecolare di NF1, tuttavia, ਠresa difficile da diversi limiti quali le dimensioni del gene, differenti tipi di mutazione e la reale assenza di una correlazione tra tipo di mutazione e decorso clinico della malattia. Fa eccezione la ricerca delle microdelezioni che sembrano essere associate ad un fenotipo pi๠grave ed ad un maggiore rischio di tumori. Il ricorso alla diagnosi prenatale, ad oggi, ਠlimitato dall'imprevedibilità del fenotipo essendo la NF1 una malattia genetica ad espressione molto variabile, soprattutto interfamiliare. NF1: diagnosi differenziale. Le macchie caffਠlatte sono un'evenienza non rara nel bambino (circa il 10-20% dei soggetti sani), spesso sono un reperto occasionale e possono non rappresentare alcun pericolo per il bambino ma, in altri casi, possono essere associate a malattie importanti come la NF1 o altre condizioni genetiche che entrano in diagnosi differenziale con essa (Tab.2) (34). Esistono altre forme, probabili varianti alleliche della NF1, nelle quali clinicamente non si hanno tutte le manifestazioni della NF1 e geneticamente, all'interno dei gruppi familiari sinora studiati, sono state riscontrate mutazioni del gene NF1 in percentuali molto variabili. Le forme "e;alleliche"e; della NF1 sono: "e;¢ la Sindrome di Watson: forma autosomica dominante caratterizzata da macchie caffਠlatte, bassa statura, deficit cognitivo e stenosi dell'arteria polmonare; "e;¢ la forma con "e;macchie caffਠlatte a trasmissione autosomica dominante"e;: caratterizzata dalla presenza di sole macchie caffਠlatte a trasmissione familiare; "e;¢ la Sindrome neurofibromatosi/Noonan: manifestazioni sovrapposte di sindrome di Noonan e NF1. La sindrome di Noonan ਠuna sindrome autosomica dominante, ad espressione variabile, dovuta a mutazioni a carico del gene PTPN11 (12q24.2-q24.31). E' caratterizzata da dismorfismi facciali, bassa statura e difetti cardiaci congeniti. La Noonan/NF1 sembra essere associata maggiormente a mutazioni del gene NF1, meno a quelle del gene PTPN11 (35). Tab. 2: Diagnosi differenziale della NF1 (da R. E Ferner. European Journal of Human Genetics (2007) 15, 131-138, modificata)
Negli ultimi anni ਠstata identificata una nuova condizione, autosomica dominante, associata alla NF1 definita Sindrome di Legius (36), dovuta a mutazioni del gene SPRED1 localizzato sul cromosoma 15 (15q13.2) (Fig.12). Essa e' caratterizzata da un fenotipo simile a quello della NF1 ma senza neurofibromi (37). Fig. 12: Localizzazione del gene SPRED1 (15q13.2), gene malattia della Sindrome di Legius
La neurofibromatosi di tipo 2 (NF2) ਠuna malattia ereditaria che si trasmette in modo autosomico dominante. Tuttavia nella metà dei casi circa, ਠdovuta ad una nuova mutazione. La sua incidenza ਠstimata intorno a 1/25.000 (38). La variabilità all'interno delle famiglie ਠminore di quella della NF1. Il gene malattia NF2 ਠlocalizzato sul cromosoma 22q12 (Fig.13) e codifica per una proteina citoplasmatica (schwannomina), sottomembranosa, che interagisce con le proteine del citoscheletro, come l'actina ed ਠimplicata nella regolazione della crescita cellulare. A livello clinico, la NF2 puಠpresentare tre gruppi di sintomi: 1) schwannomi bilaterali multipli dei nervi cranici (in generale ਠimplicato l'VIII nervo). Il numero di tumori e la loro età di insorgenza varia da un soggetto all'altro. Altri tumori del sistema nervoso centrale, essenzialmente i meningiomi e pi๠raramente gli ependimomi, sono presenti nella metà dei pazienti; 2) schwannomi sottocutanei e neurofibromi; 3) manifestazioni oculari (opacità del cristallino), per lo pi๠presenti sin dall'infanzia (39).
Un'altra forma di neurofibromatosi ਠla Schwannomatosi che clinicamente ਠcaratterizzata dalla presenza di schwannomi multipli in assenza di altre manifestazioni cliniche della NF2. Essa puಠessere sia sporadica che familiare. Sino ad oggi ritenuta una forma allelica di NF2, dovuta quindi a mutazioni del gene NF2, da studi pi๠recenti (su popolazione) ਠemerso che il gene potrebbe essere localizzato sul cromosoma 22, ma in regioni differenti da quelle della NF2 (40). Altre condizioni genetiche importanti che entrano in diagnosi differenziale con la NF1 sono la Sindrome di McCune -Albright e la Sindrome di LEOPARD. La prima ਠdefinita dai seguenti segni clinici: macchie caffਠlatte, che insorgono generalmente nel periodo neonatale, a bordi irregolari, di grandi dimensioni, localizzate prevalentemente a livello dei glutei, del sacro e della colonna vertebrale; aree multiple di displasia fibrosa e pubertà precoce. Possono presentarsi anche altre endocrinopatie da iperfunzione, che comprendono l'ipertiroidismo, l'iperincrezione dell'ormone della crescita, la sindrome di Cushing e la perdita di fosfato con le urine. La malattia ਠcausata dalle mutazioni somatiche del gene GNAS, localizzato sul cromosoma 20 (20q13.1) (Fig.14-15) ed in particolare della proteina che regola l'AMP ciclico, Gs-alfa (41). La Sindrome LEOPARD (Fig. 16) ਠuna malattia rara da difetti congeniti multipli, caratterizzata soprattutto da anomalie cardiache, cutanee e facciali. LEOPARD ਠun acronimo inglese che indica i principali segni della sindrome, che comprendono lentiggini multiple, anomalie di conduzione all'ECG, ipertelorismo oculare, stenosi polmonare, genitali anomali, ritardo della crescita e sordità neurosensoriale. Altri segni comuni sono le macchie caffਠlatte, le anomalie toraciche, il criptorchirdismo, il ritardo puberale, l'ipotonia, il ritardo dello sviluppo, di solito lieve, la sordità neurosensoriale e le difficoltà dell'apprendimento. E' una malattia autosomica dominante, a penetranza completa ed espressività variabile (42). Fig. 14-15: Gene GNAS e sua localizzazione (20q13.1) e macchie caffਠlatte caratteristiche
Fig. 16: discromie cutanee nella S. di LEOPARD (da: Anna Sarkozy et al. Orphanet Journal of Rare Diseases 2008) Diverse sono, dunque, le condizioni che entrano in diagnosi differenziale con la NF1, molte delle quali presentano le macchie caffਠlatte come segno clinico caratteristico. Per tale motivo, nel momento in cui si presenta alla nostra osservazione un bambino con macchie caffਠlatte, ਠdi fondamentale importanza valutare diversi parametri quali: numero, dimensioni, margini, superficie, distribuzione, associazione con altri segni e/o sintomi, al fine di orientarci meglio sulla corretta diagnosi. NF1: nostra casistica Dal 2003 ad oggi, presso l'ambulatorio di Genetica Clinica dell'U.O.C. di Genetica ed Immunologia Pediatrica sono state poste circa 35 diagnosi di NF1. Soltanto in 8 pazienti ਠstata identificata la mutazione del gene NF1. Di alcuni pazienti l'indagine molecolare ਠancora in corso. In 2 bambine sono state identificate due varianti mai descritte in letteratura, una delle quali in paziente con NF1 e celiachia. La nostra casistica conferma le diverse nozioni che la letteratura ci propone, in quanto anche nei nostri pazienti esiste una espressività variabile, soprattutto nei casi interfamiliari, come quello di Chiara che abbiamo visto all'età di 3 mesi, epoca in cui presentava fenotipo lieve (solo macchie caffe'latte > 6) e familiarità per NF1 (madre e nonna materna) (Fig. 17a-b-c). Fig.17: Espressività variabile in un caso interfamiliare: a) nonna; b) madre; c) figlia
In altri casi, soprattutto pediatrici, la diagnosi di NF1 non ਠcosଠsemplice in quanto, nei primi anni di vita le manifestazioni cliniche della malattia sono molto sfumate e possono comprendere talora soltanto le macchie caffਠlatte. Questo comporta un attento follow up del paziente pediatrico al fine di valutare l'evoluzione delle discromie cutanee e soprattutto la comparsa di nuove macchie e di altri segni e/o sintomi degni di nota. Tuttavia ci ਠcapitato recentemente di effettuare diagnosi di NF1 in una paziente di 45 anni (Fig.18). Questo sottolinea da un lato l'attuale disinformazione su patologie genetiche come la NF1, che pur se conosciuta risulta essere sottostimata e pi๠severa di quanto siamo abituati a pensare, e dall'altro l'importanza di una diagnosi precoce necessaria per garantire al nostro paziente una migliore qualità di vita. Fig.18: Diagnosi tardiva di NF1 in pz di 45 anni
Bibliografia 1) Minciacchi D. e Gainotti G. Malattie del Sistema Nervoso. Piccin editore. 2005 2) M. Ruggieri. Neurofibromatosi. Neurol Sci (2004) 25:S181-S183. 3) Karl McKeever et al. An epidemiological, clinical and genetic survey of Neurofibromatosis type 1 in children under sixteen years of age. Ulster Med J 2008; 77 (3) 160-163 4) Ruggieri M e Tenconi R. Le Neurofibromatosi. Associazione Linfa, Lottiamo Insieme per la Neurofibromatosi- ONLUS- (2001). 5) Seizinger BR, Rouleau GA, Ozelius LJ, Lane AH, Faryniarz AG, Chao MV, et al. Genetic linkage of von Recklinghausen neurofibromatosis to the nerve growth factor receptor gene. Cell 1987;49 (5) :589-94. 6) Ganesh Diwakar et al. Neurofibromin as a regulator of melanocyte development and differentiation. Journal of Cell Science 121, 167-177, 2008 7) Oren N. Gottfried et al. Neurofibromatosis Type 1 and tumorigenesis: molecular mechanisms and therapeutic implications. Neurosurg Focus 28 (1) :E8, 2010 8) Calvieri s. e Giustini S. Neurofibromatosi di tipo 1. Piccin editore. 2004 9) M. Upadhyaya et al. An Absence of Cutaneous Neurofibromas Associated with a 3-bp Inframe Deletion in Exon 17 of the NF1 Gene (c.2970-2972 delAAT) : Evidence of a Clinically Significant NF1 Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2007;80:140-151. 10) Rosalie E Ferner. Neurofibromatosis . European Journal of Human Genetics (2007) 15, 131-138 11) National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Arch Neurol (Chicago) 1988; 45: 575- 578 12) E. Buteica et al. Genetic and clinical considerations in six cases with neurofibromatosis type 1. Romanian Journal of Morphology and Embryology 2007, 48 (3) :243-248 13) Dugoff L, Sujansky E: Neurofibromatosis type 1 and pregnancy. Am J Med Genet 1996; 66: 7- 10. 14) Huson SM, Harper PS, Compston DAS: Von Recklinghausen neurofibromatosis: clinical and population study in South East Wales. Brain 1988; 111: 55- 81. 15) Steven L. Carroll and Nancy Ratner. How Does the Schwann Cell Lineage Form Tumors in NF1? Glia. 2008 November 1; 56 (14) : 1590-1605 16) Hagel C, Zils U, Peiper M, Kluwe L, Gotthard S, Friedrich RE, Zurakowski D, von DA, Mautner VF. Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 2007;82:187-192. 17) S. Pinson. Neurofibromatosis type I. Orphanet encyclopedia. May 2002 18) Bajenaru ML, Garbow JR, Perry A et al: Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol 2005; 57: 119-127. 19) Karl McKeever et al. An epidemiological, clinical and genetic survey of Neurofibromatosis type 1 in children under sixteen years of age. Ulster Med J 2008; 77 (3) 160-163 20) North KN, Riccardi V, Samango-Sprouse C et al: Cognitive function and academic performance in neurofibromatosis. 1: Consensus statement from the Nf1 Cognitive Disorders Task Force. Neurology 1997; 48: 1121- 1127. 21) Huson SM, Harper PS, Compston DAS: Von Recklinghausen neurofibromatosis: clinical and population study in South East Wales. Brain 1988; 111: 55- 81. 22) Bognanno JR, Edwards MK, Lee TA et al: Cranial MR imaging in neurofibromatosis. Am J Radiol 1988; 151: 381- 388. 23) Kuorilehto T, Poyhonen M, Bloigu R et al: Decreased bone mineral density and content in neurofibromatosis type 1: Lowest local values are located in the load-carrying parts of the body.Osteoporosis Int 2005; 16: 928- 936. 24) Rasmussen SA, Yang Q, Friedman JM: Mortality in neurofibromatosis 1: An analysis using US death certificates. Am J Hum Genet 2001; 68: 1110- 1118 25) Friedman JM, Arbiser J, Epstein JA et al: Cardiovascular disease in neurofibromatosis. 1: A report of the Nf1 Cardiovascular Task Force. Genet Med 2003; 4: 105- 11 26) Karlyne M. Reilly. Neurofibromatosis and lessons for the war on cancer EMBO Mol Med 1, 198-200, 2009. 27) Blanchard G. et al. Usefulness of systematic brain magnetic resonance imaging in children with Neurofibromatosis type 1. Arch Pediatr. 2009 Dec;16 (12) :1527-32 28) National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Arch Neurol (Chicago) 1988; 45: 575- 578 29) Obringer AC et al. The diagnosis of Neurofibromatosis 1 in the child under the age of 6 years. Am J Dis Child; 143:717-719, 1989. 30) Friedman JM. Neurofibromatosis 1: Clinical manifestations and diagnostic criteria. J Child Neurol 2002 31) Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet 2002; 33: 2-17 32) Alessandro De Luca, Anna Buccino, Debora Gianni, Massimo Mangino, Sandra Giustini, Antonio Richetta, Luigina Divona, Stefano Calvieri, Rita Mingarelli, and Bruno Dallapiccola. NF1 Gene Analysis Based on DHPLC. Human Mutation. Mutation in Brief .582 (2003) 33) Alessandro De Luca, Annalisa Schirinzi, Anna Buccino, Irene Bottillo, Lorenzo Sinibaldi, Isabella Torrente, Angela Ciavarella, Tania Dottorini, Roberto Porciello, Sandra Giustini, Stefano Calvieri, and Bruno DallapiccolaNovel and Recurrent Mutations in the NF1 Gene in Italian Patients with Neurofibromatosis Type 1. HUMAN MUTATION Mutation in Brief .716 (2004) 34) Rosalie E Ferner. Neurofibromatosis . European Journal of Human Genetics (2007) 15, 131-138 35) Nystr¨om AM et al. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin Genet 2009: 76: 524-534 36) Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K. Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.-P.; Cools, J.; Marynen, P.; Thomas, G.; Yoshimura, A.; Legius, E. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. (Letter) Nature Genet. 39: 1120-1126, 2007. 37) Spurlock, G.; Bennett, E.; Chuzhanova, N.; Thomas, N.; Jim, H.-P.; Side, L.; Davies, S.; Haan, E.; Kerr, B.; Huson, S. M.; Upadhyaya, M. SPRED1 mutations (Legius syndrome) : another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J. Med. Genet. 46: 431-437, 2009. 38) Asthagiri, A. R.; Parry, D. M.; Butman, J. A.; Kim, H. J.; Tsilou, E. T.; Zhuang, Z.; Lonser, R. R. Neurofibromatosis type 2. Lancet 373: 1974-1986, 2009. 39) D Gareth R Evans. Neurofibromatosis type 2 (NF2) : A clinical and molecular review. Orphanet Journal of Rare Diseases 2009, 4:16 doi:10.1186/1750-1172-4-16 40) M. Ruggieri. Neurofibromatosi. Neurol Sci (2004) 25:S181-S183. 41) Claudia E Dumitrescu and Michael T Collins. McCune-Albright syndrome. Orphanet Journal of Rare Diseases 2008, 3:12 doi:10.1186/1750-1172-3-12 42) Anna Sarkozy, Maria Cristina Digilio and Bruno Dallapiccola. Leopard syndrome. Orphanet Journal of Rare Diseases 2008, 3:13 doi:10.1186/1750-1172-3-13
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Original articles
Correlazione fra valori sierici di Icam-1 e stress ossidativo in pazienti con β-talassemia Major Correlation between serum ICAM-1 and oxidative stress in patients with β-thalassemia Major
Maria Amorini, Valeria Ferraà¹, Chiara Di Bella, Vincenzo Procopio, Giuseppina Lo Giudice, Daniela Petronilla Romeo, Alessia Micalizzi, Raffaella Mallamaci, Antonino Randazzo, Basilia Piraino, Luciana Rigoli Dipartimento di Scienze Pediatriche, UOC di Genetica ed Immunologia Pediatrica, Università degli studi di Messina
Risultati e conclusioni Nei pazienti talassemici, splenectomizzati e non, sono stati evidenziati livelli plasmatici di sICAM-1 significativamente piu' elevati rispetto ai controlli (285 ± 20 ng/mL vs. 202 ± 16 ng/mL, rispettivamente, P = 0.002).
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Analisi molecolare dei toll-like gene receptors in pazienti affetti da β-talassemia Molecular analysis of toll-like receptor gene in patients with β-thalassemia
Giuseppina Lo Giudice, Maria Amorini, Petronilla Romeo, Alessia Micalizzi, Francesca Pugliatti, Chiara Di Bella, Vincenzo Procopio, Valeria Ferraà¹, Federica Sancetta, Vincenzo Salpietro, Simona Cara, Luciana Rigoli Dipartimento di Scienze Pediatriche, UOC di Genetica ed Immunologia Pediatrica, Università degli studi di Messina
L'analisi statistica ਠstata effettata tramite il programma STATA 9 Risultati Nessuna differenza significativa ਠstata riscontrata tra la frequenza dell'allele 299Gly nel gruppo dei pazienti affetti da β-talassemia major (4, 9 %) e la frequenza nel gruppo di controllo (4, 8 %) (p=n.s.). Il medesimo risultato ਠstato ottenuto per la frequenza dell'allele 399Ile (6, 3% nei controlli, 6, 1% nei pazienti talassemici, p=n.s.). Nessun soggetto omozigote per D299G o per T399I ਠstato individuato in questo studio. Su 70 pazienti : 64 wild-type per D299G, 6 eterozigoti Su 70 controlli : 65 wild-type per D299G e 5 eterozigoti Su 70 pazienti: 65 wild-type per T399I e 5 eterozigoti Su 70 controlli: 66 wild-type per T399I e 4 eterozigoti
Conclusioni I dati del nostro studio non confermano l'ipotesi secondo la quale alcune mutazioni del gene TLR4 possono determinare una significativa alterazione della risposta infiammatoria e quindi conferire una maggiore suscettibilità alle infezioni in pazienti talassemici.
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Case reports
Una nuova mutazione del gene CYP21A2 (N387K) con interessamento di un residuo aminoacidico non conservato nell'esone 9 Novel mutation of CYP21A2 gene (n387k) affecting a non-conserved amino acid residue in exon 9
Silvestro Mirabelli, Sara Bombaci, Immacolata Rulli, Tommaso Aversa, Maria Rosa Velletri, Sabrina Cardile, Christian Freno, Filippo De Luca Dipartimento di Scienze Pediatriche, UOC di Pediatria, Università degli studi di Messina
Conclusioni a) la mutazione N387K deve essere aggiunta alla lista di mutazioni che devono essere analizzate nei pazienti con CAH; b) il genotipo V281L/N387K dovrebbe essere incluso nel gruppo di genotipi associati alle forme NC di CAH. Bibliografia 1. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21: 245-91. 2. Wasniewska M, Di Pasquale G, Rulli I, et al. In Sicilian ethnic group nonclassical congenital adrenal hyperplasia is frequently associated with a very mild genotype. J Endocrinol Invest 2007, 30: 181-5. 3. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 1986, 83: 5111-5. 4. Baradaran-Heravi A, Vakili R, Robins T, et al. Three novel CYP21A2 mutations and their protein modelling in patients with classical 21-hydroxylase deficiency from northeastern Iran. Clin Endocrinol (Oxf) 2007, 67: 335-41. 5. Robins T, Carlsson J, SunnerhagenM, Wedell A, Persson B.Molecular model of human CYP21 based on mammalian CYP2C5: structural features correlate with clinical severity of mutations causing congenital adrenal hyperplasia. Mol Endocrinol 2006, 20: 2946-64. Indirizzo per la corrispondenza Professor Filippo De Luca Dipartimento di Scienze Pediatriche Mediche e Chirurgiche, Policlinico Universitario, Via consolare Valeria, 98123 Messina, Italy Tel: + +390902213157 - Fax: + +390902212143 - e-mail: filippo.deluca@unime.it
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Nuova mutazione non-senso (W22X) nel gene CYP21A2 causante iperplasia surrenalica congenita con perdita di sali in una ragazza con eterozigosi composta Novel nonsense mutation (W22X) in CYP21A2 gene causing salt-wasting congenital adrenal hyperplasia in a compound heterozygous girl
Silvestro Mirabelli, Sara Bombaci, Immacolata Rulli, Tommaso Aversa, Maria Rosa Velletri, Sabrina Cardile, Christian Freno, Filippo De Luca Dipartimento di Scienze Pediatriche, UOC di Pediatria, Università degli studi di Messina
Abstract In our study we report the case of a Sicilian girl with congenital adrenal hyperplasia with loss of salts in which we have documented the presence of a new mutation. Riassunto Riportiamo nel nostro studio il caso di una ragazza siciliana con iperplasia congenita surrenale con perdita di sali nella quale abbiamo documentato la presenza di una nuova mutazione. Keywords: iperplasia congenita surrenale, gene CYP21A2, mutazione W22X Lista abbreviazioni: CAH - iperplasia congenita surrenale, 21-OH-D - deficit di 21 idrossilasi, PCR - reazione di polimerizzazione a catena Caso clinico Le mutazioni genetiche note come causa di deficit di 21-idrossilasi (21-OH-D) nella iperplasia surrenalica congenita (CAH) comprendono sia delezioni del gene CYP21A2 sia singole sostituzioni di aminoacidi nella proteina 21-OH, che possono comportare vari gradi di compromissione dell'attività enzimatica. Lo scopo di questo articolo ਠdi segnalare una nuova supplementare mutazione CYP21A2 in una ragazza siciliana con CAH. In una ragazza siciliana con CAH con perdita di sali diagnosticata durante le prime settimane di vita a causa di virilizzazione dei genitali (Prader III), sintomi e segni biochimici di perdita di sali ed elevati livelli sierici di 17-idrossiprogesterone (98 ng / ml) ਠstata individuata una nuova mutazione CYP21A2. Dal momento che non sono state trovate le mutazioni pi๠comuni del gene CYP21A2 che spiegherebbero il fenotipo CAH di questa paziente (1), in conformità con la nostra strategia metodologica (2, 3), ਠstato completamente sequenziato il gene CYP21A2. L'amplificazione del gene CYP21 ਠstata effettuata utilizzando primer specifici per il gene CYP21A2 che creano 3 frammenti parzialmente sovrapposti (P1P2, P3P4, CD) come altrove descritto (4). Per l'amplificazione del frammento CD, ਠstata utilizzata una Tac polimerasi (Epicentre, Madison, Wi) con un tampone privo di KCl per evitare il già descritto fenomeno dell'allele drop-out sul nucleotide in posizione 656 (5). I frammenti di PCR purificati sono stati sequenziati utilizzando il kit Big Dye Terminator Sequencing (Applied Biosystems, Warrington, Regno Unito) e primers specifici per sequenziare l'intero gene compresa la regione prossimale del promotore fino al nucleotide -370. La presenza di una larga delezione/conversione genica ਠstata esclusa dall'eterozigosi nei polimorfismi intragenici o da mutazioni e dall'analisi della segregazione degli alleli. La numerazione dei nucleotidi e degli aminoacidi segue la sequenza di riferimento di White et al. (6). Le sequenze del gene CYP21A2 nella nostra paziente hanno rivelato una sostituzione aminoacidica guanosina"e;adenina (G"e;A), che cambia il codone 22, codificante W, in un codone di stop TAG (Fig.1). Questa mutazione (W22X) ਠstata ereditata dalla madre, mentre il suo allele paterno portava la ben nota mutazione intron 2 splice, che in soggetti omozigoti ਠassociata alla forma classica della CAH (7). La conseguenza funzionale di questa nuova mutazione ਠla terminazione prematura della traduzione nel residuo 22, che porta alla produzione di una proteina tronca mancante della maggior parte dei suoi 495 aminoacidi, tra cui il residuo C429 che porta il sito di legame per l'eme, che ਠfortemente conservato in tutti gli enzimi citocromo P450. La combinazione della mutazione W22X e della mutazione intron splice 2 correla bene con il fenotipo con grave perdita di sali della nostra paziente. Una mutazione simile alla W22X (W23X secondo la nomenclatura svedese) era stata precedentemente segnalata una sola volta (http://www.cypalleles.ki.se/cyp21A2.htm). In tal caso, tuttavia, il quadro molecolare era diverso rispetto a quello rilevato nella nostra paziente: TGG"e; TGA in quel caso, (8) TGG"e; TAG nel nostro caso. In entrambi i casi questa mutazione ਠstata associata ad un fenotipo classico di CAH, ma il fenotipo era meno grave nel paziente riportato da Lajic e Wedell: una forma virilizzante semplice (8). In quel soggetto, tuttavia, il secondo allele presentava una mutazione differente: la mutazione I173N già descritta in letteratura, che ਠassociata con la forma virilizzante semplice della CAH (9).
Conclusioni Il caso da noi riportato conferma che mutazioni simili possono essere responsabili di diversi quadri molecolari e che, quando una mutazione nota non puಠspiegare pienamente il fenotipo di un paziente, il sequenziamento diretto di tutto il DNA del gene CYP21A2 ਠessenziale per trovare una nuova mutazione. Bibliografia 1. Barbaro M, Baldazzi L, Balsamo A, et al. Functional studies of two novel and two rare mutations in the 21-hydroxylase gene. J Mol Med 2006, 84: 521-8. 2. Balsamo A, Wasniewska M, Di Pasquale G, et al. Birth length and weight in congenital adrenal hyperplasia according to the different phenotypes. Eur J Pediatr 2006, 165: 380-3. 3. Di Pasquale G, Wasniewska M, Caruso M, et al. Salt wasting phenotype in a compound heterozygous girl with P482S mutation associated with a novel mutation of CYP21 gene (Q481P). J Endocrinol Invest 2005, 28: 1038-9. 4. Balsamo A, Cacciari E, Baldazzi L, et al. CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia-Romagna region. Clin Endocrinol (Oxf) 2000, 53: 117-25. 5. Day DJ, Speiser PW, Schulze E, et al. Identification of nonamplifying CYP21 genes when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital adrenal hyperplasia (CAH) affected pedigrees. Hum Mol Genet 1996, 5:2039-48. 6. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 1986, 83:5111-5. 7. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21:245-91. 8. Lajic S, Wedell A. An intron 1 splice mutation and a nonsense mutation (W23X) in CYP21 causing severe congenital adrenal hyperplasia. Hum Genet 1996, 98:182-4. 9. Wedell A, Thilà¨n A, Ritzen EM, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab 1994, 78: 1145-52. Indirizzo per la corrispondenza Professor Filippo De Luca Dipartimento di Scienze Pediatriche Mediche e Chirurgiche, Policlinico Universitario, Via consolare Valeria, 98123 Messina, Italy Tel: + +390902213157 - Fax: + +390902212143 - e-mail: filippo.deluca@unime.it
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Fenotipo con perdita di sali in una ragazza con eterozigosi composta con la mutazione P482S in associazione con una nuova mutazione del gene CYP21 (Q481P) Salt wasting phenotype in a compound heterozygous girl with P482S mutation associated with a novel mutation of CYP21 gene (Q481P)
Silvestro Mirabelli, Sara Bombaci, Immacolata Rulli, Tommaso Aversa, Sabrina Cardile, Maria Rosa Velletri, Christian Freno, Filippo De Luca Dipartimento di Scienze Pediatriche, UOC di Pediatria, Università degli studi di Messina
Abstract We report a new molecular framework in a child with the classic form of congenital adrenal hyperplasia, in which we documented the presence of the mutation P482S. Riassunto Riportiamo un nuovo quadro molecolare in una bambina con forma classica di iperplasia congenita surrenale, nella quale abbiamo documentato la presenza della mutazione P482S. Keywords: iperplasia surrenalica congenita, gene CYP21A2, mutazione P482S Lista abbreviazioni: CAH - iperplasia congenita surrenale, NC-CAH - iperplasia congenita surrenale non classica, 21-OH-D - deficit di 21 idrossilasi, K - potassio, 17OHP - 17-idrossiprogesterone, PCR - reazione di polimerizzazione a catena, ACTH - ormone adrenocorticotropo, SW - perdita di sali Caso clinico Le mutazioni genetiche note come causa di deficit di 21-idrossilasi (21-OH-D) nella iperplasia surrenalica congenita (CAH) comprendono sia delezioni del gene CYP21A2 sia singole sostituzioni di aminoacidi nella proteina 21-OH, che possono comportare vari gradi di compromissione dell'attività enzimatica. Recentemente (1) ਠstata segnalata nel codone 482 una sostituzione della prolina con la serina (P"e;S) in 7 pazienti con la forma non classica (NC) della CAH o in stato di eterozigosi; tale mutazione ਠin grado di ridurre l'attività enzimatica a circa 70% e gli autori hanno concluso che la mutazione P482S dovrebbe essere aggiunta alla lista di mutazioni che devono essere analizzate nei casi in cui sono sospettate le forme pi๠blande di CAH, soprattutto in soggetti di origine italiana. In letteratura, dopo tale segnalazione, non sono stati descritti altri casi con la stessa mutazione. Qui segnaliamo un altro paziente italiano con la stessa mutazione, che ha presentato un quadro clinico molto grave di CAH con perdita di sali (SW), a differenza dei casi riportati da Barbaro et al. (1). La diagnosi di CAH classica nella nostra paziente era stata già sospettata alla nascita a causa della grave virilizzazione dei genitali (Prader IV) ed ਠstata confermata il 2 ° giorno di vita attraverso le valutazioni biochimiche che hanno mostrato elevati livelli di K (8, 7 mmol / l), 17-idrossiprogesterone (17OHP; 102 ng / ml) e Î4-androstenedione (10, 8 ng/ml). L'amplificazione del gene CYP21 ਠstata effettuata utilizzando primer CYP21A2 specifici che creano 3 frammenti parzialmente sovrapposti (P1P2, P3P4, CD) come altrove descritto (4). Per l'amplificazione del frammento CD, e' stata utilizzata una Tac polimerasi (Epicentre, Madison, Wi) con un tampone privo di KCl per evitare il già descritto fenomeno dell'allele drop-out sul nucleotide in posizione 656 (5). I frammenti di PCR purificati sono stati sequenziati utilizzando il kit Big Dye Terminator Sequencing (Applied Biosystems, Warrington, Regno Unito) e primers specifici per sequenziare l'intero gene, compresa la regione prossimale del promotore fino al nucleotide -370. La presenza di una larga delezione/conversione genica ਠstata esclusa dall'eterozigosi nei polimorfismi intragenici o da mutazioni e dall'analisi della segregazione degli alleli. La numerazione dei nucleotidi e degli aminoacidi segue la sequenza di riferimento di White et al. (4). Dall'analisi del DNA ਠrisultato che il nostro paziente ha un allele (di origine materna) con una sostituzione aminoacidica citosina"e;timina (C"e;T), che ha causato la sostituzione di una P con una S nel codone 482 (P482S). Nell'altro allele (di origine paterna), l'analisi del DNA ha rivelato la presenza della mutazione severa intron 2 splice, che ਠcorrelata ad una forma classica di CAH in soggetti omozigoti (5). L'associazione di queste 2 mutazioni in un quadro genotipico di eterozigosi composta ਠstata precedentemente osservata solo in 1 ragazza con un fenotipo CAH molto lieve la quale ਠstata esaminata per la prima volta all'età di 17 anni per irregolarità mestruali in assenza di segni di virilizzazione (1). In quel caso, la risposta del 17OHP all'ACTH ਠstata nel limite basso del range della NC CAH (1). Oltre a queste 2 mutazioni, tuttavia, il nostro paziente presentava una mutazione supplementare, che non era mai stata descritta finora: nel filamento materno, in continuità con la mutazione P482S, la nostra ragazza presentava una sostituzione aminoacidica adenina "e;citosina (A"e;C) che ha causato la sostituzione, nel codone 481, di una glutamina (Q) con una prolina (P) (Q481P) (Fig. 1). La presenza di questa nuova mutazione nel nostro paziente puಠprobabilmente essere responsabile di una ulteriore, drammatica riduzione dell'attività della 21-OH rispetto alla singola mutazione valutata nel paziente descritto da Barbaro et al. (1), che garantiva il 70% della normale attività enzimatica arbitrariamente definita come 100%. Infatti generalmente ਠconsiderata necessaria una riduzione severa <1% per lo sviluppo di un quadro clinico di SW (5). La coesistenza di 2 mutazioni (P482S e Q481P) sullo stesso filamento puಠprobabilmente portare ad una grave compromissione dell'attività enzimatica e, quindi, essere alla base dell'espressione fenotipica di CAH osservata nella nostra ragazza.
Conclusioni a) in associazione con altre mutazioni, in codoni vicini sullo stesso filamento, la mutazione P482S puo' essere responsabile di una forma severa di CAH; b) la mutazione Q481P dovrebbe essere aggiunta alla lista di mutazioni che devono essere analizzate nei pazienti con CAH. Bibliografia 1. Barbaro M, Lajic S, Baldazzi L, et al. Functional analysis of two recurrent amino acid substitutions in the CYP21gene from Italian patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab 2004, 89: 2402-7. 2. Balsamo A, Cacciari E, Baldazzi L, et al. CYP21 analisis and phenotype/genotype relationship in the screened popula¬tion of the Italian Emilia-Romagna region. Clin Endocrinol (Oxf) 2000, 53: 117-25. 3. Day DJ, Speiser PW, Schulze E, et al. Identification of non-amplifying CYP21 genes when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital adrenal hyperplasia (CAH) affected pedigrees. Hum Mol Genet 1996, 5: 2039-48. 4. White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proc Natl Acad Sci USA 1986, 83: 5111-5. 5. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21: 245-91. Indirizzo per la corrispondenza Professor Filippo De Luca Dipartimento di Scienze Pediatriche Mediche e Chirurgiche, Policlinico Universitario, Via consolare Valeria, 98123 Messina, Italy Tel: + +390902213157 - Fax: + +390902212143 - e-mail: filippo.deluca@unime.it
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Giant Arachnoid Granulation (GAG) in a child with acute headache
Valeria Ferraà¹, Piera Vicchio, Italia Loddo, Basilia Piraino, Romina Gallizzi Department of Pediatric Sciences, UOC Pediatric Genetics and Immunology, University of Messina
Introduction The cerebrospinal fluid is normally reabsorbed by arachnoid villi or granulations of Pacchioni that allow the flow in the venous sinuses, especially in the superior sagittal sinus. Only the largest Pacchioni granulations, however, come directly into the lumen of the sinuses, while the smaller extend across the subdural space (1). We refer to arachnoid granulations as "e;giant"e; (GAG) when they have such dimensions as to fill the lumen of the venous sinus and cause local dilatation and filling defects (Kan et al, 2). Rarely, this condition causes clinical symptoms, especially a clinical picture suggestive of intracranial hypertension, however, this founding implies serious problems of differential diagnosis with other pathological conditions (cerebral venous thrombosis, neoplasms, etc.) (1). We present a case of a 10 years old patient with progressive headache and a GAG involving his transverse venous sinus. Case report A 10 year-old boy came to our observation for the second episode of acute headache associated with fever and left deviation of buccal rhyme. Physical examination appeared to be normal. The neurological and neuropsychological assessments excluded the presence of focal signs. Routinely blood tests and coagulation profile study resulted to be normal. Neuro-radiological investigations (CT, MRI and Angio-CT) showed, in the context of the gulf of right jugular, an ovoid 14.8 x 7.4 mm mass, not clearly interpretable (arachnoid granulation?, intraluminal thrombus?, glomus tumor?). Suspecting a cerebral venous thrombosis, antithrombotic treatment (enoxaparin sodium) was started, with regression of clinical symptoms. A central nervous system-MRI scan was performed again after few days of treatment. It demonstrated the filling defect without showing significant changes in the size of the lesion, thus the diagnosis of giant arachnoid granulation was confirmed. The mass was peripherally iso-intense and centrally hypo-intense to brain parenchyma with focal central calcifications. The thrombophilia tests showed an homozygosis for the MTHFR gene C677T polymorphism, with normal levels of serum homocysteine. The child, however, started folic acid therapy. Discussion Arachnoid granulations or villi are growths of arachnoid membrane into the dural sinuses, through which cerebrospinal fluid (CSF) enters the venous system from the subarachnoid space. The growth of arachnoid membrane into the dural sinus was first described by Pacchioni in 1705 (3). These projections are called arachnoid villi or arachnoid granulation (AG), depending on their size. Arachnoid villi are microscopic, whereas granulation is visible to the naked eye. AGs are absent at birth and develop in infants at the time of closure of the fontanels and increase with age, in numbers and size, in response to increased CSF pressure from the subarachnoid space and are usually quite evident by 4 years of age (4). AGs, in general are a rare finding with a reported prevalence of 0.3-1% in the adult population, especially in the elderly (aged > 65 years). Arachnoid granulations are most commonly seen at the junction between the middle and lateral thirds of the transverse sinuses (90%), near the entry sites of the superficial veins; they are also detectable, in decreasing frequency, in the cavernous sinus, superior petrosal sinus and straight sinus (2). They normally measure a few millimetres, but they may grow sufficiently to partially occlude and enlarge the dural sinus. AGs are considerable "e;giant"e; when they are of sufficient size to fill the lumen of a dural sinus and cause local dilation or filling defects (2, 4). They rarely cause symptoms of increased intracranial pressure from venous hypertension secondary to partial sinus occlusion. If symptoms are present, intra-sinus pressure measurements across the lesion through each dural sinus can be used to determine whether the lesion is causing venous outflow obstruction and venous hypertension. The complete physiological role of AG and the mechanism of the formation aren't well understood and still controversial. Current theories argue the arachnoid villi function as a passive filtration system for cerebrospinal fluid, providing a pathway from the subarachnoid space into the venous system (2, 5). In the differential diagnosis of masses within the dural sinus there are: meningiomas, arachnoid cystis, dermoid cystis and venous sinus thrombosis. It's crucial to exclude the latter since is potentially dangerous and needs immediate management (5, 6). Cranial CT and MRI can be used to distinguish both conditions (7, 8). In literature has been described only a case, occurred in pediatric age, similar to that one reported by us, in which the diagnostic hypothesis of venous sinus thrombosis was initially taken in account and promptly excluded (9). In our patient, the tests for thrombophilia have shown an homozygosis for the MTHFR gene C677T polymorphism, with normal levels of serum homocysteine. MTHFR is an enzyme involved in homocysteine metabolism. Homozygotes have more than 50% reduced enzyme activity, but the effect of impaired MTHFR function on homocysteine levels is dependent on folate intake (10). The classic MTHFR C677T gene polymorphism is weakly associated with an increased risk of VTE (Venous thromboembolism). The relative risk for venous thromboembolism, due to MTHFR decreased activity, is slightly increased when a condition of double heterozygosis is present. However, our patient started folic acid therapy. We experienced a case of a giant arachnoid granulation misdiagnosed as dural sinus thrombosis. Before diagnosing the sinus thrombosis, giant arachnoid granulation should be considered in differential diagnosis. MRI is the most useful tool to differentiate giant arachnoid granulation from dural sinus thrombosis. Fig 1: Angio-CT finding
References 1. CB Grossman, DG Potts Arachnoid granulations: radiology and anatomy Radiology 1974 113: 95-100 2. Kan P, Stevens EA, Couldwell WT et al. Incidental giant arachnoid granulation AJNR 2006 27:1491-1492 3. LeGros Clark WE: o the pacchionian bodies. J Anat 55:40-48, 1920 4. S.A. Peters, E. Frombach, CM Heyer Giant arachnoid granulation: differential diagnosis of acute headache Eur Radiol 2006 19 :1046 5. Chang Won Cho Giant arachnoid granulation misdiagnosed as transverse sinus thrombosis Radiology 2002 105:333-341 6. Y. Kiroglu, B. Yagci, B. Cirak et al. Giant arachnoid granulation in a patient with benign intracranial hypertension Eur Radiol 2009 19 :1046 7. T. Koshikawa, S. Naganawa, H. Fukatsu et al. Arachnoid Granulations on high-resolution MR images and diffusion-weighted MR images: normal appearance and frequency Radiation Medicine 2007 18:187-191 8. Mamourian AC, Towfighi J rt al. MR of giant arachnoid granulation: a normal variant presenting as a mass within the dural venous sinus Am J neuroradiol 1995; 16:901-04 9. S.C.Chin, C.Y. Chen, C.C. Lee et al. Giant arachnoid granulation mimicking dural sinus thrombosis in a boy with headache: MRI Neuroradiology 1998 40:181-183 10. I. Bezemer, MS Carine, M Doggen, et al. No association between the common MTHFR 677C T polymorphism and venous thrombosis Arch Intern Med 2007 11. M. Den Heijer, S. Lewington, R. Clarke Homocysteine, MTHFR and risk of venous thrombosis : a meta-analysis of published epidemiological studies Journal of Thrombosis and Haemostasis 2005;3:292-9
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
Editorial letter
Rachitismo Ipofosfatemico
C. Moscheo, V. Salpietro Damiano, G. Stroscio*, S. Cardile, M. Biasi**, S. Mazziotti* UOC Pediatria, Dipartimento di Scienze Pediatriche Mediche e Chirurgiche *UOC Radiodiagnostica, Dipartimento di Scienze Neurologiche, Messina **O.O.R.R. Bianchi-Melacrino-Morelli Reggio Calabria
Gentile direttore, descriviamo il caso di un bambino di 5 anni giunto per la prima volta alla nostra osservazione all'età di 2 anni e 9/12 per accertamenti diagnostici in merito ad una sospetta"e; osteodisplasia"e;. Familiarità positiva per rachitismo ipofosfatemico e bassa statura disarmonica. Unicogenito, nato a termine da gravidanza fisiologica esitata in parto eutocico. Fenomeni neonatali nella norma. Nulla da segnalare fino all'età di 14 mesi quando, in coincidenza dell'avvio della deambulazione, il piccolo ha presentato un incurvamento in varismo degli arti inferiori associato ad una progressiva deflessione della curva di crescita staturale. Per un inquadramento diagnostico sono state eseguite radiografie di tibia, perone, radio e ulna , che hanno dimostrato la presenza di modificazioni strutturali. La zona dia-epifisaria si presentava sfrangiata con metafisi deformata a coppa ed incurvamento della diafisi (quadro radiografico che depone per osteodisplasia), (Fig. 1). L'esame ematochimico documentava la presenza di normocalcemia, ipofosfatemia, incremento dei livelli ematici di fosfatasi alcalina e livelli ematici di PTH nella norma. Inviato alla nostra osservazione per tale quadro il piccolo veniva sottoposto a valutazione clinica che evidenziava la presenza di tibie a sciabola con varismo importante degli arti inferiori (Fig. 2), lordosi lombare, andatura anserina, riscontro di braccialetto rachitico. E' stata rilevata inoltre presenza di displasia dello smalto dentario (Fig. 3). Sono stati ripetuti esami di laboratorio che hanno confermato la presenza di normocalcemia, aumento dei livelli ematici di fosfatasi alcalina, ed ipofosfatemia con iperfosfaturia. In considerazione della familiarità , dell'esito delle indagini eseguite su sangue ed urine ed in base al riscontro radiografico delle alterazioni scheletriche ਠstata dunque posta diagnosi di rachitismo ipofosfatemico familiare e avviata terapia con fosfato e vitamina D3. Discussione Il rachitismo ipofosfatemico X-linked identifica una condizione geneticamente determinata caratterizzata dalla presenza di un'alterazione dei tubuli prossimali renali (XLH) cui consegue un patologico controllo del metabolismo calcio-fosforo-vitamina D con perdita di quest'ultima attraverso l'emuntorio renale (OMIM 307800). La sua incidenza ਠstimata essere di 1:200000 (1).La patologia si presenta in genere con un quadro di scarsa crescita (bassa statura), che si manifesta durante i primi due anni di vita , con alterazioni strutturali dell'apparato osteo-articolare (incurvamento degli arti inferiori, osteomalacia e rachitismo) e dei denti. L'esecuzione di esami di laboratorio nei pazienti affetti documenta presenza di ipofosfatemia con iperfosfaturia, normocalcemia e normali livelli ematici di paratormone .I valori di vitamina D possono essere normali o ridotti (ciಠਠimputabile a due fattori: ridotta attività della1, 25 idrossilasi e aumento dei processi ossidativi che inattivano il vitamero metabolicamente attivo della vitD(2-3). L'ipofosfatemia ਠprobabilmente il dato di laboratorio pi๠precoce, si sviluppa entro i primi mesi di vita e pertanto andrebbe ricercata in pazienti con familiarità positiva. Una caratteristica distintiva del rachitismo ipofosfatemico X-linked ਠla presenza di lesioni compatibili con una ridotta mineralizzazione ossea periosteocitica in sede corticale a livello dell'apparato scheletrico. In merito all'eziopatogenesi sono state avanzate diverse ipotesi relative ad alterazioni a carico dei geni coinvolti nella sintesi di trasportatori renali di fosfato che non sono state confermate. E' stata invece accertata, nella maggior parte dei pazienti affetti, la presenza di mutazioni del gene PHEX (4), localizzato sul braccio corto del cromosoma X, che codifica per una endopeptidasi espressa principalmente a livello di osteoblasti e odontoblasti ma non a livello renale(5). Tale prodotto pare sia coinvolto nel catabolismo dei fosfati, al punto che potrebbe prevenire il clivaggio di una fosfoglicoproteina (fosfatonina) della matrice extracellulare che interviene nel controllo dei livelli sierici del fattore FGF23(6). L'inattivazione del gene PHEX correla infatti con un aumento dei livelli circolanti di FGF23 cui conseguirebbe comparsa di successiva fosfaturia. E' stata inoltre documentata una relazione inversa tra FGF23 e riduzione dei livelli ematici di fosfato, evidenze recenti infatti indicano che elevati livelli di FGF23 giocano un ruolo centrale nel determinismo dell'ipofosfatemia interferendo con il metabolismo della vitamina D con comparsa dei segni clinici della malattia (7). E' interessante segnalare che l'affinità del FGF23 verso il suo recettore ਠbassa (8), questo sembra indicare un coinvolgimento, nella trasduzione del segnale indotta dall'interazione ligando-recettore, di altre molecole. A tal proposito in letteratura viene descritta l'interazione del fattore FGF23 con una proteina di superficie, denominata Klotho, identificata in topi transgenici nei quali la sua espressione ਠridotta. Il ruolo di Klotho nel determinismo dell'azione del fattore FGF23 e dunque nel controllo del metabolismo calcio-fosforo, ਠsuggerito da quanto segue: la proteina ਠespressa a livello renale, paratiroideo, ipofisario e del plesso corioideo (9); i livelli di FGF23 risultano essere elevati nei topi Klotho, l'iniezione di FGF23 in tali animali induce l'espressione di fattori di crescita e fenomeni di trasduzione di segnale; elementi FGF23-null e topi Klotho mostrano fenotipi caratterizzati da iperfosfatemia, livelli elevati di vit D, calcificazioni rilevate a livello dei tessuti molli e una ridotta aspettativa di vita; dati di laboratorio indicano inoltre che anticorpi antiKlotho aumentano fosfatemia e i livelli di vitamina D nei topi wilg-type, Klotho sembra necessario, come mediatore del fattore FGF23, per l'induzione della riduzione dei livelli sierici di fosfato e vitamina D. Studi recenti hanno mostrato infine che Klotho, FGF23 e FGFR1c formano in vitro un complesso in cui il legame tra FGF23 e recettore ਠpotenziato dalla proteina Klotho (10). In merito alla terapia attualmente trovano impiego somministrazione di fosfato ad alte dosi e calcitriolo (11). Benchਠla terapia sia efficace nel determinare il raggiungimento di adeguati livelli sierici di fosfato e nel correggere rachitismo ed osteomalacia ਠgravata da effetti collaterali quali nefrocalcinosi e iperparatiroidsmo secondario. Un'alternativa alla terapia tradizionale, valutata in alcuni studi, propone l'impiego di cinacalcet, un calciomimetico che agisce riducendo i livelli di PTH e incrementando la fosfatemia, il suo impiego ridurrebbe inoltre di entità il supplemento di fosfati e calcitriolo. A tale composto sembra inoltre associarsi un ridotto rischio di iperparatiroidismo secondario, ipercalcemia, ipercalciuria e nefrocalcinosi (12). Si attendono in atto ulteriori studi per poter procedere con l'eventuale utilizzo di tale sostanza nei pazienti affetti.
Bibliografia 1)Tenenhouse HS, Econs MJ (2001) Mendelian hypophosphatemias. In: Scriver CR, Beaudet AL, Sly WS, Valley D (eds). The metabolic basis of inherited disease. McGraw-Hill, New York, pp 5039-5067 2). Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004). FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429-435 3) Perwad F, Zhang MY, Tenenhouse HS, Portale AA (2007). Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am J Physiol Renal Physiol 293: 1577-1583 4) The HYP Consortium (1995). A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nature Genet 11:130-136 5) Quarles LD (2003). FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab 285:E1-E9 6) Garabedian M (2007). Regulation of phosphate homeostasis in infants, children, and adolescents, and the role of phosphatonins in this process. Curr Opin Pediatr 19:488-491 7) White KE, Larsson TE, Econs MJ (2006).The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev 27:221-241 8) I. Urakawa, Y. Yamazaki, T. Shimada, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature, vol. 444, no. 7120, pp. 770-774, 2006. 9) S. A. Li, M. Watanabe, H. Yamada, et al.. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Structure and Function, vol. 29, no. 4, pp. 91-99, 2004. 10) H. Kurosu, Y. Ogawa, M. Miyoshi, et al..Regulation of fibroblast growth factor-23 signaling by Klotho. Journal of Biological Chemistry, vol. 281, no. 10, pp. 6120-6123, 2006. 11) Baroncelli GI, Bertelloni S, Sodini F, Galli L, Vanacore T, Fiore L, Saggese G. Genetic advances, biochemical and clinical features and critical approach to treatment of patients with X-linked hypophosphatemic rickets. Pediatr Endocrinol Rev 1: 361-379, 2004 12) Uri S. Alon, Rachel Levy-Olomucki, Wayne V. Moore, Jason Stubbs, Shiguang Liu, and L. Darryl Quarles. Calcimimetics as an Adjuvant Treatment for Familial Hypophosphatemic Rickets. Clin J Am Soc Nephrol 3: 658-664, 2008
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
Rivista Italiana di Genetica e Immunologia Pediatrica - Italian Journal of Genetic and Pediatric Immunology
Anno II numero 1 - gennaio 2010 | direttore scientifico: Carmelo Salpietro - direttore responsabile: Giuseppe Micali
geneticapediatrica.it trimestrale di divulgazione scientifica dell'Euromediterranean Paediatric Foundation Legge 7 marzo 2001, n. 62 - Registro della Stampa Tribunale di Messina n. 3/09 - 11 maggio 2009
 Direttore scientifico Carmelo Salpietro
Direttore scientifico Carmelo Salpietro